Hierarchical MIL (Multiple Instance Learning) 방법론과 비교하기 위해 사용된 최신 대표 모델에 대해 알아보자.
(참고) Hier-MIML 논문 리뷰 :

🔹 ScRAT
- 기반: Transformer 기반 모델
- 핵심 아이디어: 단일 세포 표현 간의 상호작용을 주고받는 self-attention을 이용해, 각 세포 표현을 업데이트
- 장점: 세포 간 관계를 학습하여 세포 간의 영향을 잘 모델링함
- 출처: Mao et al., 2024
- 요약: attention으로 세포 간의 상호작용까지 포착하는 능력이 있음
[ScRAT] Phenotype prediction from single-cell RNA-seq data using attention-based neural networks
[ScRAT] Phenotype prediction from single-cell RNA-seq data using attention-based neural networks
논문 https://academic.oup.com/bioinformatics/article/40/2/btae067/7613064정리 Attention 기반 신경망을 사용한 / 단일 세포 RNA-Seq 데이터의 / 표현형 예측Attention 기반으로 진행하는 것이 이 논문의 핵심!Attentino
doraemin.tistory.com
🔹 ProtoCell4P
- 기반: 프로토타입 기반 신경망
- 핵심 아이디어: 세포 표현들이 특정 "prototype" 주변에 클러스터 되도록 latent 공간을 구성함
- 장점: 세포 하위 집단을 명시적으로 모델링하고 해석력 확보
- 논문: Xiong, Bekiranov, and Zhang, 2023
https://pmc.ncbi.nlm.nih.gov/articles/PMC10444962/ - 요약: "대표 세포군"을 latent space에 정의하고, 세포들이 이들과 유사하도록 유도

- 방법론 :
- First, it has a cell embedding module, which encodes the input gene expression data of a single cell into a low-dimensional latent space and learns embedded cell prototypes automatically.
- 각 단일 세포의 유전자 발현 데이터를 저차원 잠재 공간(latent space)에 인코딩하는 autoencoder 구조를 사용
- 학습 중 cell prototypes라는 세포 하위 집단(subpopulations)의 대표 벡터들을 자동으로 생성 ( 학습 과정에서 자동으로 생성된 잠재 벡터들 )
- prototype이 특정 cell type 그룹의 평균 임베딩과 가까워지도록 유도.
- cell type을 분류하는 classifier를 학습해서 prototype이 실제 cell type 구분에 도움 되도록.
- prototype이 특정 cell type 그룹의 평균 임베딩과 가까워지도록 유도.
- 세포 임베딩 :
- reconstruction loss: 입력 데이터를 잘 복원하도록
- cell-to-prototype distance 최소화: 각 세포가 적어도 하나의 프로토타입 근처에 위치하도록
- prototype 간 거리 최대화: 프로토타입이 서로 다른 하위 집단을 잘 표현하도록
- Second, it has a classification module, which classifies an individual by estimating dynamic relevance scores of prototypes for each cell and integrating the information from all cells in a sample.
- 각 세포는 자신과 프로토타입 간의 유사도(similarity) 를 계산하고, 이를 기반으로 각 프로토타입이 해당 환자 분류에 얼마나 중요한지 relevance score를 출력
- 학습된 cell embeddings과 prototype 간 유사도(s_ij)를 기반으로
- relevance scorer라는 작은 네트워크가 각 세포마다 다르게 relevance를 추정 → adaptive relevance
- 각 세포의 relevance score를 계산 (U(x_ij))
- 각 세포의 정보는 relevance와 유사도를 곱해서 weighted contribution으로 환자의 phenotype을 추론
- 각 prototype이 환자 분류에 얼마나 중요한지 학습
- 이 구조는 세포 수가 달라도 환자 단위 예측을 할 수 있다.
- 각 세포는 자신과 프로토타입 간의 유사도(similarity) 를 계산하고, 이를 기반으로 각 프로토타입이 해당 환자 분류에 얼마나 중요한지 relevance score를 출력
- First, it has a cell embedding module, which encodes the input gene expression data of a single cell into a low-dimensional latent space and learns embedded cell prototypes automatically.
+ ProtoCell4P는 CloudPred보다 더 미세하게 세포 개별의 역할을 학습하고, 해석력도 더 뛰어남을 강조.

🔹 CloudPred
- 기반: Gaussian Mixture Model (GMM)
- 핵심 아이디어: 샘플 내 세포들을 GMM으로 클러스터링한 후, 각 클러스터의 비율을 feature로 사용해 예측
- 장점: 세포 분포를 통계적으로 모델링
- 논문: He et al., 2021
https://pubmed.ncbi.nlm.nih.gov/34890161/
chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://psb.stanford.edu/psb-online/proceedings/psb22/he.pdf - 요약: 세포 분포(클러스터 비율)를 이용해 전체 샘플을 표현함
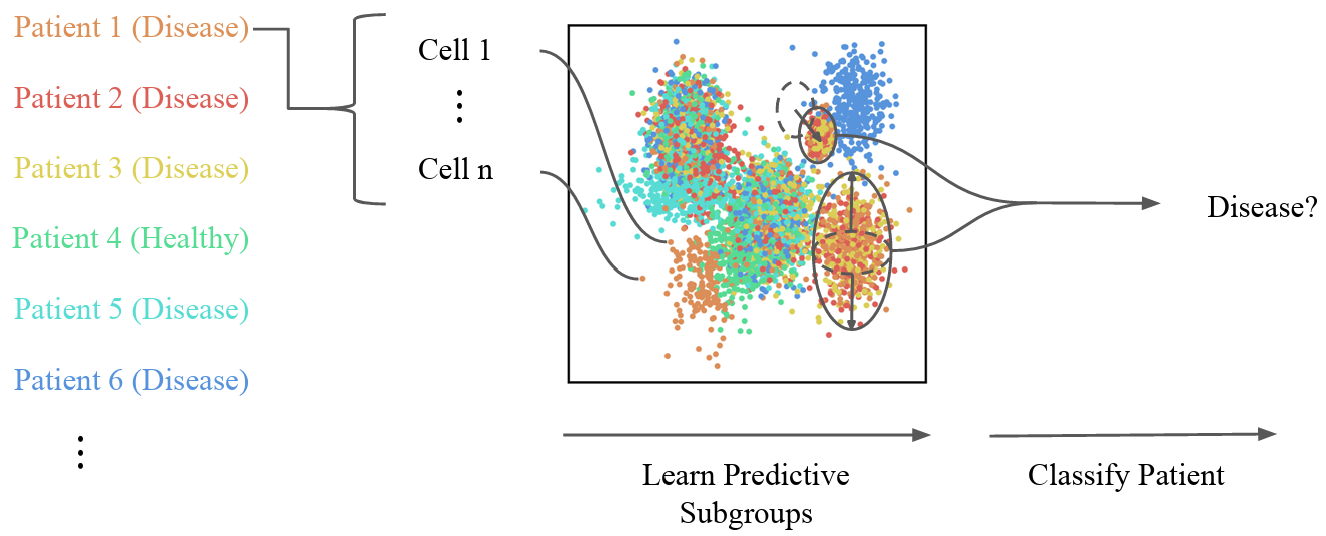
- 방법론:
- Gaussian Mixture Model (GMM):
- 모든 환자의 scRNA-seq 데이터를 포인트 클라우드(point cloud)로 간주 ( 그림 속 점 하나하나가 한 개의 세포 (cell) )
- 각 세포를 혼합 가우시안 모델(GMM)을 통해 특정 서브타입으로 분류

- 모든 세포들을 m개의 클러스터로 나누어, 개의 Gaussian 분포로 세포군(subpopulation)을 설명
- 각 세포는 soft하게, 즉 확률적으로 각 클러스터에 속함
- 세포 x_i가 클러스터 j에 속할 확률 : p_ij 계산
- 모든 환자 간에 공통된 서브타입(클러스터 중심)을 학습함.
- 클러스터 별 가중치:
- 세포가 각 클러스터에 속할 확률을 계산
- 환자 P의 세포들을 클러스터별로 정리해서 고정된 길이의 벡터 s∈{R}^m 생성
- 각 클러스터의 상대적인 abundance를 벡터로 구성함.
- : 클러스터 j에 속할 확률의 평균 → 세포 집합 내 클러스터 j의 abundance
- 세포가 각 클러스터에 속할 확률을 계산
- 표현형 분류기:
- abundance 벡터를 입력으로 받아 표현형을 예측하는 2차식(quadratic) 모델을 사용.
- 예측은 softmax 또는 logistic regression을 통해 이루어짐.
- Gaussian Mixture Model (GMM):
+ 정리 비교
| 방식 | GMM 정의 방식 | 장점 | 단점 | 사용 상황 |
| CloudPred | 전체 환자의 세포로 공통된 m개 Gaussian 학습 | 클러스터 의미 해석 가능 (모든 환자에서 공통 서브타입) , 예측 정밀도 높음 | 계산량은 많음 | 환자 간 세포 수, 세포 다양성 모두 고려하고 싶음 |
| Mixture (patient) | 환자별로 따로 GMM 학습 | 환자별 특이성에 민감하게 작동 (세포 패턴이 독특한 환자에게 유리) | 표현형 예측용 classifier가 generalize되지 않음 (클러스터 해석 불가), 공통된 클러스터가 없어서 비교 불가능 | 세포 수 적고 빠르게 대조군/질병군을 비교하고 싶음 |
| Mixture (class) | 질병/정상 별로 따로 GMM 학습 | 표현형 구분에 도움되는 대표 세포 분포를 학습, 약간의 해석 가능성 있음 (루푸스 클러스터 vs 정상 클러스터) | 환자 간 다양성 간과 (개별 환자들의 세포 분포 차이를 무시), 미세한 세포 간 차이나 상호작용을 포착하기 어려움 | 개별 환자 분석 위주, 개인 맞춤적 탐색 |

🔹 MixMIL
- 기반: Attention MIL + Generalized Linear Mixed Model (GLMM)
- 핵심 아이디어: attention을 이용해 sample-level representation을 만들고, 그 후 GLMM을 통해 예측
- 장점: noise에 robust하고, MIL과 통계 모델링의 장점 결합
- 논문: Engelmann et al., 2024
https://proceedings.mlr.press/v238/engelmann24a/engelmann24a.pdf - 요약: attention으로 feature를 추출하고 GLMM으로 해석력과 견고함 확보

- 방법론 :
- Attention 기반 Bag Embedding
- 하나의 bag X∈{R}^{I \times Q}은 I개의 instance (e.g., 세포)를 포함하고, 각 instance는 Q-차원의 임베딩 벡터 x_i를 가짐.
-
더보기
🧬 예시
Bag (사람) Instance (세포) 세포 표현 (Q=3인 임베딩) 환자 A (X) 세포 1 (x₁) [0.2, -1.3, 0.7] 세포 2 (x₂) [0.0, 0.4, 0.1] 세포 3 (x₃) [1.1, -0.5, 2.0] ... ... ... - 각 bag은 하나의 label 을 갖지만, 개별 instance에 대한 label은 없음.
-
- MixMIL은 복잡한 deep network 대신, 선형 attention layer로 중요도를 계산
-
더보기

MixMIL이 일부러 선형으로 만든 이유
- 계산 효율성 (빠르고, 파라미터 적음)
- 과적합 방지 (특히 데이터가 작을 때)
- 해석 용이성 (어떤 feature가 중요했는지 추적 가능)
- Bag Embedding 계산: instance들의 weighted sum, 즉 중요한 세포들의 정보를 강조한 표현

-
- 하나의 bag X∈{R}^{I \times Q}은 I개의 instance (e.g., 세포)를 포함하고, 각 instance는 Q-차원의 임베딩 벡터 x_i를 가짐.
- GLMM과의 결합
- attention으로 구성된 bag 임베딩을 GLMM의 설명 변수로 넣는 방식

-
더보기GLMM이란 :
"선형 모델"을 기반으로 하되,
고정 효과(fixed effect)와 랜덤 효과(random effect)를 모두 반영하는 모델
용어 뜻 예시 선형 모델 y=Xβ+ϵy = X\beta + \epsilon 키 = 2 × 몸무게 + 10 GLM (Generalized Linear Model) 선형 모델 + 다른 분포 (e.g. 분류) 예: 로지스틱 회귀 Mixed Model 고정 효과 + 랜덤 효과 사람마다 반응이 다를 수 있음 GLMM GLM + Mixed Model 분류 + 그룹 차이까지 반영! 
예: 학교별로 학생의 시험 성적 예측
- 고정 효과: 공부시간 → 모든 학생에게 동일하게 작용
- 랜덤 효과: 학교마다 다르게 작용하는 요인 → 학교별 intercept 다르게 설정
왜 쓰는가?
장점 설명 개인차 반영 사람, 샘플, 배치 등마다 달라지는 효과 모델링 가능 해석 가능 어떤 요인이 얼마나 영향을 주었는지 분리 가능 MIL과 궁합 attention으로 만들어진 embedding을 잘 받아줌 다양한 분포 대응 이진 분류 (Bernoulli), 다중 분류 (Categorical), 연속값 예측까지 가능
- attention으로 구성된 bag 임베딩을 GLMM의 설명 변수로 넣는 방식
- Attention 기반 Bag Embedding
Hierarchical MIL vs. MixMIL
| 항목 | Hierarchical MIL | MixMIL |
| 구조적 특징 | sample > group > instance의 계층적 구조 모델링 | 단일 수준의 MIL 구조를 사용하되, attention weight와 GLMM을 결합 |
| 세포 간 의존성 | 중첩된 구조에서 다양한 수준의 정보 고려 | 세포 간 중요도를 weight로 표현, 간접적으로 heterogeneity 반영 |
| 임베딩 처리 | 자체 학습 or feature 학습 필요 | 사전학습(pretrained)의 instance embedding 사용 (e.g. scVI, SimCLR) |
| 모델링 방식 | 딥러닝 중심 | 통계적 GLMM + MIL attention 결합 (shallow 구조) |
| 해석 가능성 | 일부 가능 | 높음: 예측이 instance-level 영향의 가중합으로 표현됨 |
| 복잡도 | 높을 수 있음 (특히 deep HMIL) | 매우 낮음 – 단일 linear layer로 attention, efficient training |
ScRAT vs. MixMIL 구조적 차이 설명:
1. Attention Mechanism의 목적이 다름
- ScRAT은 self-attention을 통해 셀 간의 관계(correlation)를 파악해 transformer-like 표현 학습을 한다.
- MixMIL은 attention을 통해 각 셀의 중요도(weight)를 학습하고, 이를 이용해 bag-level 임베딩을 만든다.
2. Embedding 처리 방식
- ScRAT은 raw scRNA-seq 데이터를 attention layer를 통해 end-to-end로 처리한다.
- MixMIL은 사전 학습된 cell embedding (X)을 입력으로 받아 shallow attention layer (linear + softmax)만 학습한다.
3. 해석성
- ScRAT은 high-attention cell을 이용해 phenotype-driving cell population을 도출한다.
- MixMIL은 attention-weighted instance embedding을 GLMM의 해석 가능한 선형 predictor로 사용하는 구조라 통계적 해석이 더 용이하다 (zᵀβ = ωᵀXβ 형태로 설명 가능).
+ 비교

'AI & Data Analysis' 카테고리의 다른 글
| [scMILD] Datasets Download (0) | 2025.04.03 |
|---|---|
| Difference btw '.h5ad' and '.h5' (0) | 2025.03.28 |
| [Alzheimer] 녹음 파일 불러와서 이미지로 변환하기 (0) | 2025.03.23 |
| [ScRAT] STEP 1. Sample mixup (0) | 2025.03.20 |
| [SAM] 모델 다운로드 및 실행 (0) | 2025.02.26 |


