PAPER
https://arxiv.org/abs/2208.03325
Isoform Function Prediction Using a Deep Neural Network
Isoforms are mRNAs produced from the same gene site in the phenomenon called Alternative Splicing. Studies have shown that more than 95% of human multi-exon genes have undergone alternative splicing. Although there are few changes in mRNA sequence, They ma
arxiv.org
초록(Abstract)
연구의 배경(Alternative Splicing으로 생성되는 isoform의 기능적 중요성)과 문제의식(isoform 기능에 대한 실험적 정보 부족)을 제시합니다.
기존 MIL 기반 방법들이 갖는 한계(라벨 부족, 성능 미흡)와 본 연구의 기여(시퀀스·발현·GO 정보 통합, Semi-supervised DNN 모델)를 간결히 요약합니다.
label은 오로지 gene(유전자) 단위로만 주어져 있습니다.
따라서 gene을 하나의 bag(가방)으로 보고, 그 안에 속한 여러 isoform을 instance(인스턴스)로 간주합니다.
모델은 각 isoform에 대해 예측 점수(“이 isoform이 해당 기능을 수행할 가능성”)를 출력하고,
그 isoform 점수들 중 최댓값(max pooling) 을 해당 gene의 예측 점수로 삼아,
gene-level 라벨(실제 기능 유무)과 비교하며 학습합니다.
이 구조 덕분에 “isoform별로 라벨이 없더라도” gene-level 라벨만으로 isoform 기능 예측이 가능합니다.
1. 어떤 isoform이 그 기능(label)에 가장 크게 기여하는지
모델은 각 isoform마다 “이 기능을 수행할 확률”에 해당하는 점수를 내놓습니다. 그중 최댓값을 gene-level 예측에 사용하므로, 최종적으로 “가장 높은 점수를 받은 isoform”이 어떤 것인지 알 수 있습니다. 이를 통해 해당 유전자가 그 기능을 가지게 하는 주요 isoform(들)을 식별할 수 있죠.
2. gene-level label 예측
isoform 점수들 중 가장 높은 하나만으로 gene-level label을 예측합니다. 즉, “여러 isoform의 조합”이라기보다는, “이 중 하나의 isoform이 충분히 높은 점수를 가지면 gene도 그 기능을 가진다”는 MIL 가정 하에 학습·예측이 이루어집니다. 이 논문에서는 “최댓값” 방식을 사용해, 가장 핵심적인 isoform 하나를 지목하고, 그 점수만으로 gene-level label을 결정하도록 한 것이 특징입니다.
서론(Introduction, 1장)
DNA→RNA→단백질의 중심원리와 Alternative Splicing의 메커니즘을 그림(그림 1)과 함께 설명하며, 동일 유전자에서 생성되는 isoform이 기능적으로 서로 다를 수 있음을 강조합니다.
- Isoform이란?
Isoform(아이소폼)은 하나의 유전자에서 Alternative Splicing 과정을 통해 생성되는 서로 다른 형태의 mRNA(그리고 단백질) 변형체를 가리킵니다.- 유전자는 연속된 DNA 염기서열 중에서 Exon(코딩 영역)과 Intron(비코딩 영역)으로 구성됩니다.
- 전사(transcription) 과정에서 Intron은 잘려나가고 Exon만이 mRNA로 이어집니다.
- 이때 “어떤 Exon을 포함시키고, 어떤 Exon을 제외시킬지”의 조합이 달라지면, 동일한 유전자에서 서로 다른 mRNA(아이소폼)가 만들어집니다.
- 이렇게 생성된 서로 다른 mRNA를 번역하면, 구조나 기능이 약간씩 다른 단백질이 나오게 되는데, 이를 isoform이라 부릅니다.
대표적인 예시 : BCL2L1 유전자는 BCL-XS(소형)와 BCL-XL(대형) 두 가지 아이소폼을 만드는데,
BCL-XS는 세포자멸사(apoptosis, 세포 사멸)를 촉진하는 반면,
BCL-XL은 세포자멸사를 억제합니다.

BCL2L1, CASP3 등의 사례를 통해 “isoform별 기능 예측의 필요성”을 제시하고, 기존 iMILP·WLRM·DeepIsoFun·DIFFUSE 등의 한계를 언급하며 본 연구의 방향을 명확히 합니다.
재료 및 방법(Materials and Methods, 2장)
1. 데이터셋(2.1절)
19,303개 유전자로부터 39,375개 isoform, 1,735개 실험의 발현 프로파일, UniProt GO(Graph 구조를 갖는 4,184개 용어) 등 입력·출력 데이터를 구체적으로 설명합니다.
39,375개 isoform 각각에 대해, “실제로 각 실험 샘플에서 이 isoform이 얼마나 발현되었는지”를 측정하기 위해 334개의 서로 다른 RNA-Seq 연구(study)에서 총 1,735개의 개별 시퀀싱 실험(experiment)의 발현 데이터를 수집해 사용한 것입니다.
UniProt Gene Ontology(GO) 데이터베이스는 생물학적 기능을 계층적·그래프 구조로 조직해 놓은 표준 사전입니다.
각 유전자는 이미 알려진 기능에 따라 하나 이상의 GO 용어에 매핑(mapping)되어 있습니다.
본 연구에서는 총 4,184개의 GO 용어를 사용해, 19,303개 유전자의 기능 레이블을 구성했습니다
2. 단백질 시퀀스 임베딩(2.2.1절)
동적 임베딩의 파라미터 폭발 문제를 지적하고, 사전학습된 Protvec를 활용하는 이유와 보존 도메인(domain)에는 trainable embedding을 사용함을 기술합니다.
- 단백질 서열(sequence)을 신경망에 입력하기 위해서는 먼저 숫자 벡터로 변환하는 임베딩(embedding) 과정을 거쳐야 합니다. Isoform 논문 2.2.1절에서는 이 임베딩을 두 가지 방식으로 구분하여 설명합니다.
Static 임베딩(Pre-trained embedding)
미리 대규모 데이터로 학습된 고정된 벡터를 사용합니다. 학습 과정에서 업데이트되지 않기 때문에 파라미터 수가 늘지 않고, 데이터가 적어도 과적합 위험이 낮습니다.
Dynamic 임베딩(Trainable embedding)
모델 학습 과정 중에 함께 업데이트되는 임베딩입니다. 문제 특이적 특징을 잘 학습해 성능을 높일 수 있지만, 임베딩 차원이 크고 학습 데이터가 부족하면 과적합이 심해질 수 있습니다 .
- 단백질 서열에 대한 Dynamic 임베딩의 문제점
전형적인 dynamic 임베딩은 3-mer(amino-acid 3글자 조합) 단위로 서열을 분할하여 벡터화합니다.
아미노산 종류가 20개이므로 가능한 3-mer 조합은 20³ = 8,000개.
Isoform 논문에서 평균 단백질 길이는 약 3,000이므로, 임베딩 파라미터 수는 8,000 × 3,000 = 24백만 개에 달합니다.
이렇게 파라미터 수가 커지면 학습 데이터(≈39,375 isoform × 1,735 실험)가 충분치 않아 심각한 과적합을 초래할 수 있습니다 .
- Protvec: 사전학습된 Static 임베딩의 채택
동적 임베딩의 과적합 문제를 피하기 위해, 논문에서는 Protvec라는 사전학습(pre-trained) 임베딩을 사용합니다.
Protvec는 대규모 단백질 서열 코퍼스에서 미리 학습된 3-mer 임베딩으로, 고정된 벡터를 제공합니다.
이로써 모델 파라미터를 크게 늘리지 않으면서도, 단백질 서열의 연관성을 잘 포착할 수 있습니다 .
- 보존 도메인(conserved domain)에 대한 Dynamic 임베딩
단백질 서열 전체와 달리, 보존 도메인의 종류는 약 16,000개로 상대적으로 적습니다.
따라서 도메인 서열에 대해서는 학습 과정 중에 업데이트되는 dynamic 임베딩을 사용해도 파라미터 폭발 위험이 적고, 문제 특이적 표현을 학습할 수 있다고 판단하여 trainable embedding을 적용합니다.
이렇게 Isoform 논문은
- 단백질 서열 전체 → 사전학습된 Protvec(static embedding)
- 보존 도메인 → Dynamic embedding(trainable)
을 조합하여, 과적합은 억제하면서도 단백질 기능 예측에 필요한 표현력을 확보하도록 설계하고 있습니다.
보존 도메인이란?
Isoform: 하나의 유전자가 alternative splicing으로 만들어 내는 서로 다른 전체 단백질 변형체(transcript/peptide).
Conserved domain: 그 단백질(isoform) 안에 포함된, 기능적으로 의미 있는 부분 서열.
---
보존 도메인(conserved domain)은 단백질 내에서 기능적으로나 구조적으로 보존되어 온 짧은 서열 단위입니다.
예를 들어, 단백질-단백질 상호작용, DNA 결합, 효소 활성 부위 등 특정 기능을 수행하는 도메인들이 여기에 해당하며, 일반적으로 Pfam, CDD(Conserved Domain Database) 같은 데이터베이스에서 정의됩니다. 논문에서는 이 보존 도메인 항목을 약 16,000종으로 한정하여, 각 isoform이 갖는 도메인들의 순서를 LSTM으로 처리하는 dynamic embedding을 적용함으로써, 문제 특이적 표현을 학습하도록 설계했습니다
3. 제안 모델 구조(2.2.2절, 그림 3·4)
(1) Protvec + CNN을 통한 단백질 서열(sequence) 특징 추출
입력: 각 isoform의 아미노산 서열
처리:
사전학습된 Protvec로 3-mer 단위 임베딩을 수행하여 고차원 벡터 생성
이 임베딩 벡터를 1D CNN 레이어와 이후 Dense 레이어에 통과시켜, 시퀀스 기반의 compact feature vector를 추출
출력: 시퀀스 특징 벡터.
1D CNN 사용 이유? (아래 접은글 참고)
CNN은 이미지 처리에 자주 쓰이지만, 본 논문에서 쓰인 것은 1D CNN으로, 시퀀스 데이터(여기서는 Protvec 임베딩된 3-mer 벡터 시퀀스)에 적합한 구조입니다. 그 이유를 정리하면 다음과 같습니다.
- 국소적 패턴 추출
- 단백질 서열에는 특정 아미노산 조합(모티프)이 중요한 기능적 의미를 갖습니다. 1D CNN의 필터(커널)는 이처럼 연속된 3-mer 임베딩 벡터들 사이의 국소적 패턴(local motif) 을 자동으로 학습해낼 수 있습니다 .
- 파라미터 공유 및 효율성
- CNN 필터는 시퀀스 전체에 걸쳐 동일한 가중치를 공유(translation invariance)하므로, 서열 길이가 길어도 과도한 파라미터 증가 없이 특징을 학습할 수 있습니다.
- 이는 RNN 계열보다 학습이 빠르고, 병렬 처리에도 유리합니다.
- 차원 축소 및 잡음 억제
- CNN 뒤에 이어지는 풀링(pooling) 또는 dense 레이어는 중요한 특징을 compact feature vector로 요약하면서, 시퀀스 전반의 잡음을 줄여 줍니다 .
- 다른 도메인에서도 검증된 방법론
- 텍스트(단어 임베딩)나 음성(스펙트로그램) 처리에서도 1D/2D CNN이 널리 쓰이며, 단백질 서열 분석 분야에서도 DeepGO 같은 선행 연구가 CNN 기반 시퀀스 특징 추출의 유효성을 입증했습니다.
따라서, “숫자로 된 벡터”더라도 그 벡터들이 갖는 연속적이고 구조적인 패턴을 효과적으로 포착하기 위해 1D CNN을 활용하는 것이며, 이는 이미지 외의 다양한 시계열·서열 데이터에 적용 가능한 범용적인 기법입니다.

(2) 보존 도메인(conserved domain) 순서 정보 학습 (Dynamic embedding + LSTM)
입력: 각 isoform 서열에서 검출된 보존 도메인의 리스트(약 16,000종 중 해당 isoform에 존재하는 순서대로)
처리:
도메인 종류별로 학습 가능한 trainable embedding을 적용
이 임베딩 시퀀스를 LSTM 모듈에 통과시켜, 도메인이 나타나는 순서(order) 정보를 캡처
출력: 도메인 기반 특징 벡터.

(3) 발현 프로파일(expression profile) 특징 학습
입력: RNA-Seq 실험에서 얻은 isoform별 정규화된 발현값(334 studies · 1,735 experiments 기반)
처리:
노이즈 제거를 위한 정규화(normalization) 컴포넌트 적용
이어서 여러 개의 Dense 레이어를 거쳐, 발현 패턴을 요약한 expression feature vector 추출
출력: 발현 기반 특징 벡터.

(4) 이 세 특징 벡터를 결합하여 isoform 수준 스코어를 예측
시퀀스, 도메인, 발현으로부터 얻은 3개의 feature vector를 concatenate
다시 Dense 레이어를 지나며 통합된 isoform-level score(각 GO term에 대한 점수)로 변환
이 과정을 통해 isoform 하나당 다중 속성을 반영한 예측치를 생성

(5) MIL( Multiple-Instance Learning )의 max-pooling을 응용해 isoform→유전자 스코어로 변환
- 배경: 라벨(기능) 정보는 gene 수준으로만 제공
- 연산: 하나의 gene에 속한 모든 isoform 점수 중 최댓값(max pooling) 을 해당 gene의 점수로 취함
- 이유: “gene이 특정 기능을 가지려면, 그 기능을 수행하는 isoform이 최소 하나는 있어야 한다”는 MIL 가정에 부합
As a result, the customized max-pool is designed to get the maximum score of the gene’s isoforms and assign it to the specific gene. By applying this component, the scores are transferred from the isoform level to the gene level, and the training process can be performed.
문제 설정
Bag(가방): 라벨(기능) 정보가 알려진 단위, 본 논문에서는 “gene”이 bag이 됩니다.
Instance(인스턴스): 라벨이 없는 작은 단위, 여기서는 하나의 gene이 가질 수 있는 여러 “isoform”이 instance가 됩니다.
MIL 가정
“어떤 기능을 가진 gene이라면, 그 gene에 속한 최소한 하나의 isoform(instance)이 해당 기능을 수행할 것이다.”
반대로, gene이 기능이 없다고 하면, 그 gene의 어느 isoform도 그 기능을 수행하지 않는다고 간주합니다.
Max-Pooling 연산
각 isoform에 대해 예측된 기능 점수들 중 최댓값(max score) 을 해당 gene의 점수로 취합니다.
이를 통해 isoform 수준 예측을 gene 수준 라벨과 맞춰 학습할 수 있습니다
이처럼, MIL은 “bag에만 라벨이 있고, instance에는 라벨이 없는” 약지도 학습(weakly supervised learning) 기법으로, 본 연구에서는 gene-level 기능 라벨을 활용해 isoform-level 예측을 가능하게 해 줍니다.
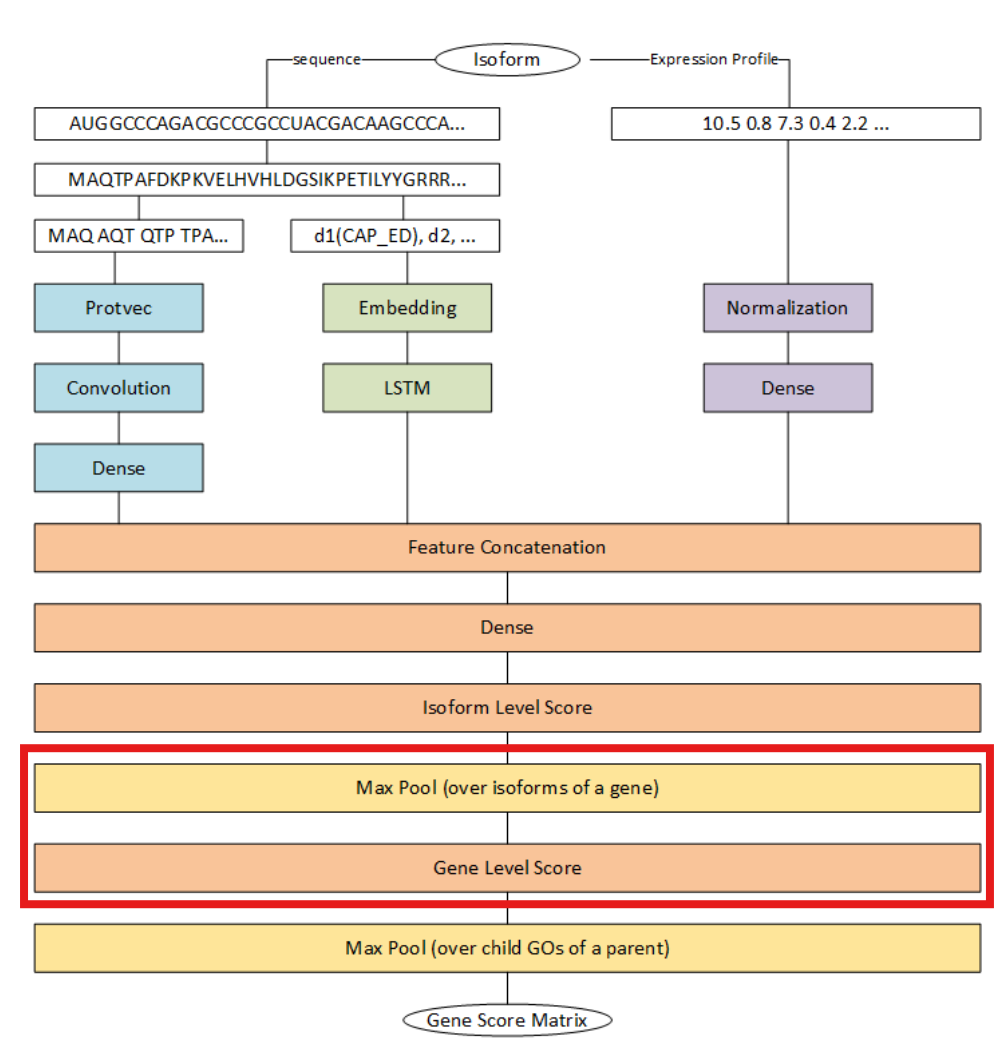
(6) GO 계층구조 반영(max-pooling over hierarchy)
- 입력: Gene-level score 행렬
- 연산: GO 그래프에서 자식(child) 용어의 점수와 부모(parent) 용어의 점수를 비교해 더 큰 값을 부모에 할당
- 효과: “하위 기능이 존재하면 상위 기능도 동시에 가질 것”이라는 생물학적 계층구조를 모델에 주입

각 isoform에 대해 “어떤 GO term을 예측할지”는 결국 모델의 최종 출력층(output layer) 구조로 결정됩니다. 구체적으로 단계별로 살펴보면: (아래 접은 글)
- 특징 벡터 합치기
- 앞서 설명한 대로, Protvec+CNN → domain LSTM → expression Dense 로부터 얻은 3개의 특징벡터를 concatenate 해서 하나의 통합된 isoform-level 특징벡터 hh를 만듭니다.
2. GO term 수만큼의 출력 유닛
- 논문에서는 4,184개의 GO term을 예측 대상으로 삼기 때문에, 이 통합 벡터 hh를 입력으로 받는 Dense layer (또는 여러 개의 Dense layer) 의 마지막에 4,184개의 뉴런을 배치합니다.
- 이 마지막 레이어의 가중치 W∈Rd×4184W\in\mathbb{R}^{d\times 4184}, 편향 b∈R4184b\in\mathbb{R}^{4184} 를 통해 각 GO term에 대응하는 logit 벡터를 계산합니다.
- ${logits} = hW + b$
- 3. 활성화 함수로 확률화
- 각 term별로 “이 isoform이 이 기능을 수행할 확률”을 얻기 위해 sigmoid 함수를 적용합니다.
- $s_i = \sigma(\text{logit}_i)\quad(i=1,\dots,4184)$
- 이 $s\in[0,1]^{4184}$ 벡터가 “isoform-level GO term 점수”가 됩니다.
4. 학습 과정에서의 지도(signal)
- isoform에는 직접 라벨이 없으므로, gene-level 라벨과 MIL 구조를 통해 간접적으로 이 출력벡터를 학습합니다.
- isoform별 ss 벡터 → gene별 max-pooling → gene-level 예측벡터 gg
- $g$ 벡터와 실제 gene-level GO 라벨 간의 다중 이진분류 손실을 계산
- 역전파로 $W,b$ 포함 모델 전체 파라미터를 업데이트
- 이렇게 학습된 $W,b$가 “어떤 특징이 어떤 GO term과 연관이 있다”를 자동으로 학습하여, isoform $h$가 주어졌을 때 각 term의 점수를 매기도록 만듭니다.
정리하면,
- 매칭은 “특징벡터 → (4,184) 차원 출력 → sigmoid” 의 구조로,
- 각 출력 유닛 하나하나는 특정 GO term에 대응하며,
- 학습 과정에서 MIL과 multi-task 손실에 의해 그 유닛이 해당 term과 연관된 특징을 포착하도록 가중치가 조정됩니다.
4. 학습 기법(2.2.3절)
Multi-task learning 관점에서 4,000개 GO를 한 번에 학습하기 위한 샘플링 전략과 Dynamic Weight Average를 이용한 손실 가중치 자동 조정 방식을 설명합니다.
결과(Results, 3장)
성능 비교: Dataset 1·2·3에 대해 AUC·AUPRC 지표로 iMILP, WLRM, DeepIsoFun, DIFFUSE와 비교. 제안 기법이 복잡도는 낮추면서 성능은 경쟁력이 있음을 보입니다 (Table 1) .
구성 요소 중요도 분석: GO 계층 반영과 Protvec 교체 실험을 통해 각각 성능이 얼마나 떨어지는지 제시하여, 제안 요소의 기여도를 정량적으로 평가합니다 .
isoform 수준 검증: 문헌에 기능이 보고된 14개 isoform 중 10개를 정확히 예측하여 정성적 타당성을 보충합니다 (Table 2) .
결론(Conclusion, 4장)
본 연구가 “단일 네트워크로 GO 전체를 학습”함으로써 연산 비용과 과적합 위험을 줄이면서도 높은 예측 정확도를 확보했음을 요약합니다.
향후 연구로는 발현 데이터의 GCN 모델링, MTL 심화 기법(특화 층 증가·클래스 클러스터링 후 학습) 등을 제안합니다 .



