[PAPER] RECOVERING TIME-VARYING NETWORKS FROM SINGLE-CELLDATA
https://arxiv.org/abs/2410.01853
Recovering Time-Varying Networks From Single-Cell Data
Gene regulation is a dynamic process that underlies all aspects of human development, disease response, and other key biological processes. The reconstruction of temporal gene regulatory networks has conventionally relied on regression analysis, graphical
arxiv.org
2. MEHODS
2.1 PROBLEM SETUP
- X는 $cell*gene$ Matrix 형태.
- gene은 25,000~35,000
- cell은 수천개~수백만개
- Dynamic Graph에서 cell에 time이 있다고 가정해보자.
- 그럼 $X_t $ 는 $cell_t * gene$ 형태
- $cell_t$는 $t$마다 달라질 수 있음
- 그럼 $X_t $ 는 $cell_t * gene$ 형태
- Directed graph $\widetilde{G} := {G1, ... , G_T}$를 복원하는 것이 목표이다.
- graph node는 gene의 집합. $N=[g]$ ; static
- graph edge는 $\varepsilon_t = {(u,v,x)}$; dynamic
- v : target
- u : target v의 발현을 조절하는 gene
- w : 이 관계의 강도
- source node를 전사 인자 유전자(TF)라고 합니다.
- $ G_t $를 $A_t$로 특성화하자.
- $ \widetilde{X} = f ( \widetilde{X} ^ {TF}, \widetilde{A} ) $
- $ \widetilde{X} ^ {TF} $ 는 모든 $TFs$의 발현을 나타냄
- f는 복잡해서 알려져있지 않음
- f를 autoencoders로 학습하기도 함
- 하지만 모든 gene expression을 reconstruction 하기엔 어렵다.
- $ \widetilde{X} = f ( \widetilde{X} ^ {TF}, \widetilde{A} ) $
$ \widetilde{X} $ 는 원래 데이터로 우리가 가진 input입니다.
그러나 여기서 이 수식은:
"$\widetilde{X}_{\text{TF}}$ 와 모델이 학습한 $\widetilde{A}$으로 전체 발현 $\widetilde{X}$를 복원할 수 있는지 보는 reconstructive insight"입니다.
즉, 이건 "생물학적으로 f라는 함수가 존재한다고 가정"하는 것이고, 실제로는 Marlene은 이걸 직접 복원하려 하기보다는, 이를 활용해서 cell type 분류로 우회 학습합니다.

- GRNs는 cell function에 의존한다. => cell type 예측 문제로 단순화해보자!
- 이는 GRNs 복구도 가능하게 할 것이다.
- 분류 문제 $y = f ( \widetilde{x} ^{TF} , \widetilde{A} )$
- $ \widetilde{x} ^ {TF}$ : 동일한 유형의 세포로 구성된 시간적 배치가 주어졌을 때
- $ \widetilde{x} ^ {TF} ∈ ℝ ^ {T×배치 크기×n}$
- y는 정답 cell type

2.2 ARCHITECTURE OF MARLENE
Marlene은 세 가지 주요 단계로 구성됩니다.
처음 두 단계는 (1)에서 h의 선택을 다루고,
마지막 단계는 f의 선택을 다룹니다.

< first step : 유전자 특징화 단계 >
- input : $X$ (cell*gene)
- method : PMA (Pooling by Multihead Attention) ; “세포” 위에서 작동 ; PMA는 attention을 "세포들을 요약하는 통계"로 활용
- output : $G = H^T$ (gene*k feature)
- k : 모델이 학습하고자 하는 '출력 통계 벡터의 수', 즉 "pooling summary vector의 개수"를 의미. PMA가 학습한 집합의 요약 통계(vector summary)를 나타냄
* 하나의 batch에는 -> 하나의 cell type만 데이터로 주어져야한다!
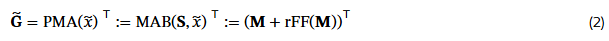
| 단계 | 연산 이름 | 적용대상 | 목적 | Query/Key/Value | 구조 출력 | 비유 |
| 1단계 | PMA (Pooling by Multihead Attention) | **세포(Cell)**들 | 각 유전자에 대한 통계 요약 벡터 생성 | Query: 학습 가능한 Seed vector $S$ Key/Value: $X \in \mathbb{R}^{c \times g}$ | $H \in \mathbb{R}^{k \times g} $ → 전치 → $G \in \mathbb{R}^{g \times k}$ | “여러 사람들(세포)의 의견을 듣고 요약하는 작업” |
| 2단계 | Self-Attention (GRN 학습용) | **유전자(Gene)**들 | 유전자 간 조절 관계 (GRN) 추론 | Query/Key: $G_t W_t^Q, G_t W_t^K $ from gene feature $G_t$ |
Adjacency matrix $A_t \in \mathbb{R}^{g \times g}$ (혹은 $g \times p$) | “요약된 전문가들(유전자)끼리 서로 대화하며 관계도를 그리는 작업” |
한줄 요약
PMA: 세포들을 정리해서 유전자별 표현을 만드는 attention → “요약 pooling”
GRN Self-Attention: 유전자 표현들 간의 관계를 모델링하는 attention → “유전자 네트워크 학습”
비유로 쉽게 이해하면?
1단계 PMA는 “여러 사람들(세포)의 의견을 듣고 요약하는 작업”
2단계 Self-Attention은 “요약된 전문가들(유전자)끼리 서로 대화하며 관계도를 그리는 작업”
< second step : self-attention >
- input : $G_t$
- method : self-attention 시행.; “유전자” 위에서 작동 ; GRN을 위한 “유전자들 사이의 상호작용( 유전자 간의 관계, 조절 방향(→) )을 모델링”하는 attention
- output : $A_t$ ; gene regulatory network, 즉 directed weighted graph
- 생물학적 타당성과 계산 효율성을 위해 열(즉, 조절자 역할을 하는 유전자)을 TF(transcription factors)로 제한하여 $A_t \in \mathbb{R}^{g \times p}$ 로 만듭니다.
- 여기서 $p$는 TRRUST 데이터베이스에 있는 알려진 TF의 수입니다.
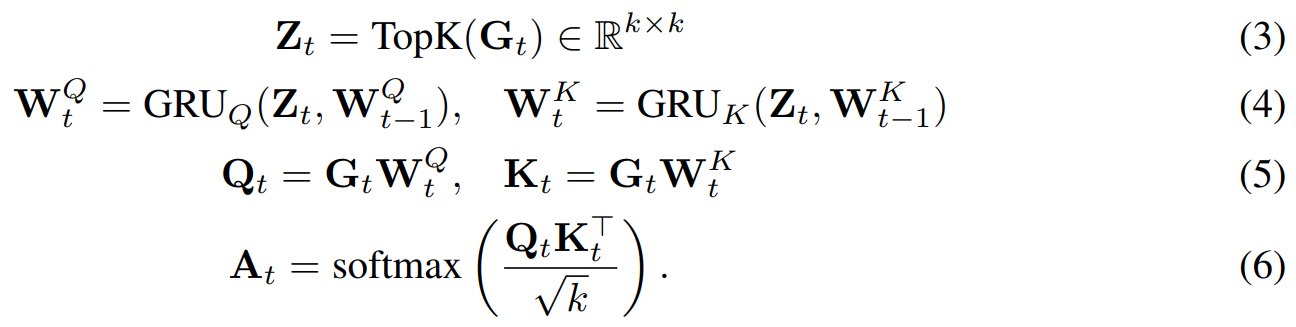
* GRU를 통해 self-attention의 Query/Key weight $(W_t^Q,W_t^K)$ 가 시간 t에 따라 변하도록 구성
즉, 이전 시간 $t−1$의 weight를 기반으로 → 현재 시간 t의 weight를 GRU를 통해 "진화(evolve)"시킴
=> W가 스스로 시간에 따라 업데이트 되도록 구성된 것!
* Since most time series we deal with contain very few time points, a GRU should suffice and not suffer from vanishing gradient problems.
우리가 다루는 대부분의 시계열은 시간 지점이 매우 적기 때문에 GRU로 충분하며 기울기 소멸 문제가 발생하지 않습니다.
→ Marlene이 다루는 scRNA-seq 시계열 데이터는 보통 시간 지점 수가 적음 (예: 4~7)
→ 따라서 복잡한 LSTM보다는 GRU가 적합
* 상위 중요 유전자 𝑘개만 뽑아 정사각 행렬 $Z_t∈R^{k×k}$ 생성 → GRU 입력으로 사용
< Last Step: Gene Expression Reconstruction & Cell Type Prediction >
- input : $ \widetilde{x} ^ {TF} \widetilde{A} ^ {T} $ → TF 유전자들의 발현값과 복원된 시간별 GRN $\widetilde{A}$를 조합한 표현
- method : 여러 개의 fully connected (FC) layer + 비선형 활성화 함수 $\sigma$
- output : $ \widetilde{y} ∈R^C $ → 총 cell type 수만큼의 logit vector
→ CrossEntropyLoss를 통해 학습

* $A_t∈R^{g×p}$ → $A_t^⊤∈R^{p×g}$
* $x ̃^{TF} A ̃^⊤ ∈ R^{cell× g} $ 즉, TF의 발현과 gene 연결 정보(GRN)를 곱해서 전체 유전자 발현값(cell type 예측)을 얻는 형태
