STEP 0. 공식 튜토리얼이 자세히 잘 나와있다. 잘 참고하자.
https://www.10xgenomics.com/support/software/cell-ranger/latest/tutorials
Cell Ranger - Official 10x Genomics Support
A set of analysis pipelines that perform sample demultiplexing, barcode processing, single cell 3' and 5' gene counting, V(D)J transcript sequence assembly and annotation, and Feature Barcode analysis from single cell data.
www.10xgenomics.com
STEP 1. cellranger-x.y.z.tar.gz 설치
https://www.10xgenomics.com/support/software/cell-ranger/latest
Cell Ranger - Official 10x Genomics Support
A set of analysis pipelines that perform sample demultiplexing, barcode processing, single cell 3' and 5' gene counting, V(D)J transcript sequence assembly and annotation, and Feature Barcode analysis from single cell data.
www.10xgenomics.com
'Download Cell Ranger'를 클릭하면, 개인 정보를 입력 후,
Download Center로 이동되어 (자신만의) 다운로드 코드가 나온다.


tar.gz 파일을 받아보자.
코드를 복사하여, linux 터미널에 입력해주었다.
curl -O cellranger-8.0.1.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-8.0.1.tar.gz?Expires=XXXX&Signature=XXXX&Key-Pair-Id=XXXX"
or
wget -O cellranger-8.0.1.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-8.0.1.tar.gz?Expires=XXXX&Signature=XXXX&Key-Pair-Id=XXXX"

다운받은 파일을 압축 해제해주자.
tar -xzvf cellranger-8.0.1.tar.gz
tar -xzvf cellranger-x.y.z.tar.gz //x.y.z 버전에 맞게 입력
(확장자가 .xz인 경우)
tar -xvf cellranger-x.y.z.tar.xz
STEP 2. reference data files 설치
Download Center에서, Human reference와 Mouse reference를 다운 받자.

Human reference 다운로드하고, 압축 해제하자.
curl -O "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2024-A.tar.gz"
tar -xzvf refdata-gex-GRCh38-2024-A.tar.gz

Mouse reference 다운로드
curl -O "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCm39-2024-A.tar.gz"
tar -xzvf refdata-gex-GRCm39-2024-A.tar.gz
+) 별도로 참조 데이터베이스 생성할 필요 없음
- refdata-gex-GRCh38-2024-A.tar.gz 파일은 10x Genomics에서 미리 생성한 데이터베이스를 제공합니다.
- 따라서, 별도로 참조 데이터베이스를 생성할 필요 없이, 바로 사용할 수 있습니다.
STEP 3. $PATH에 Cell Ranger 디렉토리를 추가
이렇게 하면 cellranger 명령을 호출할 수 있습니다.
export PATH=/경로/cellranger-8.0.1:$PATH
export PATH=/data/project/kim89/Cell_ranger/cellranger-8.0.1:$PATH
.bashrc // 편의를 위해 이 명령을 추가할 수 있습니다.
+) 잘 설치 되었는지 확인
cellranger --versionSTEP 4. Cell Ranger count를 실행해보자!
이것도 튜토리얼에 자세히 나와있다
https://www.10xgenomics.com/support/software/cell-ranger/latest/tutorials/cr-tutorial-ct
Running Cell Ranger count - Official 10x Genomics Support
A set of analysis pipelines that perform sample demultiplexing, barcode processing, single cell 3' and 5' gene counting, V(D)J transcript sequence assembly and annotation, and Feature Barcode analysis from single cell data.
www.10xgenomics.com
cellranger-8.0.1 에서 실행해야 한다.
cd 파일경로/cellranger-8.0.1cellranger count --id=sample1_analysis \
--transcriptome=/home/kim89/Cell_ranger/refdata-gex-GRCh38-2024-A \
--fastqs=/home/kim89/Cell_ranger/fastq_output/ \
--sample=SRR3879601,SRR3879602,SRR3879603 \
--localcores=8 --localmem=64- --id - A sample ID. This is used for naming the outputs
- --transcriptome - the directory containing the Cell Ranger reference
- --fastqs - the directory containing the fastq files
This will process all fastq files in the --fastqs directory into a single sample. If you have multiple samples in a single directory then you need to add:
- --sample - the SampleName from the fastq files as above.
In addition, Cell Ranger is very computationally intensive, you will usually be wanting to run it on a high performance cluster or server, and it is advisable to set limits to the resources it will use:
- --localcores - the number of processors Cell Ranger should use
- --localmem - the amount of memory, in Gigabytes, Cell Ranger should use.

+) 다른 데이터에서도 마찬가지로 cellranger count를 실행해보았다.
cellranger count --id=SRR11606920_analysis \
--transcriptome=/data/project/kim89/Cell_ranger/refdata-gex-GRCh38-2024-A \
--fastqs=/data/project/kim89/SRR116069__/ \
--sample=SRR11606920
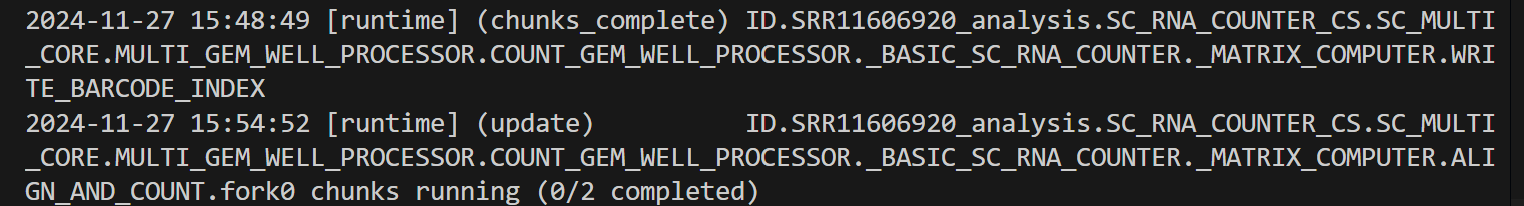



--create-bam 옵션 : BAM 파일 생성 여부를 지정합니다.
- true: BAM 파일을 생성합니다. (일반적으로 설정)
- false: BAM 파일을 생성하지 않습니다.
BAM 파일이란?
- BAM 파일(Binary Alignment/Map)은 바이너리 형태의 파일로, 시퀀싱된 리드가 참조 유전체에 정렬된 정보를 포함합니다.
- SAM 파일의 압축 버전이며, 더 작은 파일 크기와 빠른 접근 속도를 제공합니다.
- 주요 정보:
- 각 리드(read)가 참조 유전체의 어느 위치에 매핑되었는지.
- 매핑 품질 점수.
- UMI(Unique Molecular Identifier) 및 바코드 정보.
BAM 파일 생성의 장점
- 품질 검사 및 검증
- 정렬된 리드를 시각화하여 매핑 품질을 확인할 수 있습니다.
- IGV(Integrative Genomics Viewer)와 같은 도구를 통해 리드가 특정 유전자에 얼마나 잘 매핑되었는지 시각적으로 검토할 수 있습니다.
- 추가 분석 가능
- BAM 파일은 downstream 분석(예: 돌연변이 탐지, RNA 편집 분석)에서 추가 데이터 소스로 사용됩니다.
- 재분석 용이
- 이후 새로운 분석 방법을 적용할 때 정렬 데이터를 재사용할 수 있습니다.
- 원본 FASTQ 파일을 다시 처리할 필요가 없습니다.
BAM 파일 생성의 단점
- 용량 증가
- BAM 파일은 대량의 데이터를 포함하므로 디스크 공간을 더 많이 차지합니다.
- 보통 수 기가바이트(GB) 단위의 크기를 가집니다.
- 불필요할 수 있음
- 만약 단일 세포 데이터의 downstream 분석에서 BAM 파일이 필요하지 않다면, 생성하지 않아도 됩니다.
- 예를 들어, Cell Ranger는 count matrix(유전자-세포 발현 매트릭스)를 생성하는 데만 사용된다면 BAM 파일이 필수적이지 않습니다.
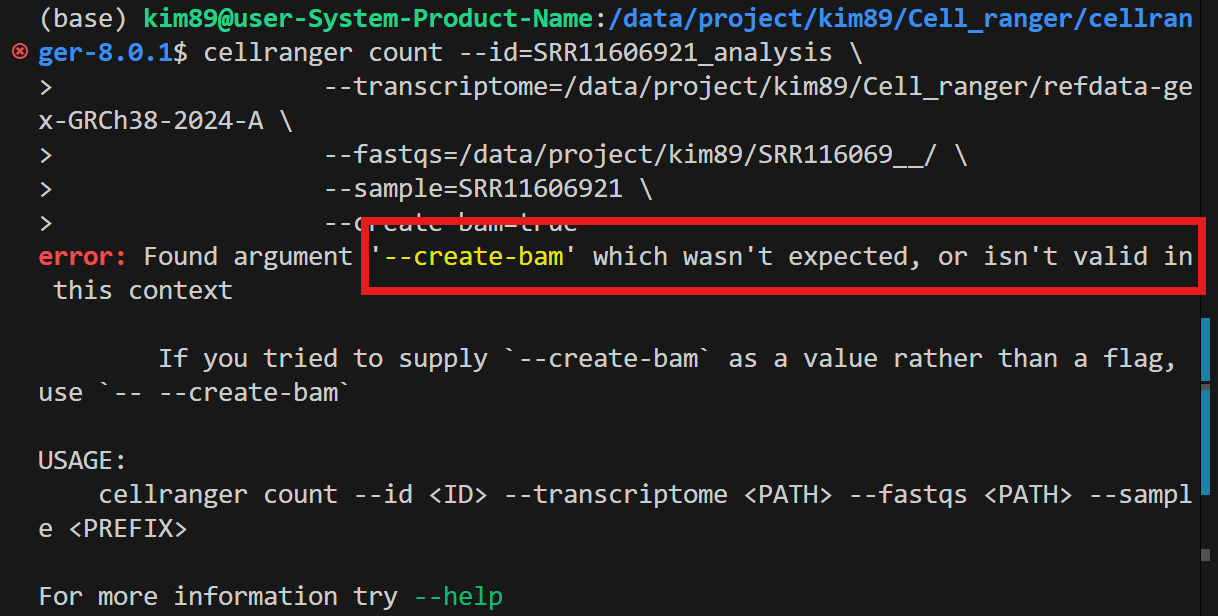
- --create-bam 옵션은 최신 Cell Ranger 8.0.1에서는 유효하지 않습니다.
- 옵션을 제거하고 실행하면 BAM 파일은 기본적으로 생성됩니다.
- 명령이 성공적으로 실행될 것입니다! 😊
+) 진행 상태 확인해보자
1. 다른 터미널을 열고 cell ranger count 작업 진행 중인 디렉토리로 이동
cd /data/project/kim89/Cell_ranger/cellranger-8.0.1/SRR11606920_analysis
2. 로그 파일 확인
less _log
3. 로그 파일 빠져나오려면 'q'를 누르면 된다.
- 오류 로그를 확인 ⇒ 오류가 없으면 이 파일은 비어 있을 수 있다.
less _errors
- 실시간 로그 확인 : 로그 파일이 업데이트될 때마다 새로운 내용이 표시된다. (실시간 로그 보기 중단하려면 'Ctrl + C')
tail -f _log- 디스크 공간 확인
df -h+) 파일 명 에러 !
(base) kim89@user-System-Product-Name:~/Cell_ranger/cellranger-8.0.1$ cellranger count --id=sample1_analysis --transcriptome=/home/kim89/Cell_ranger/refdata-gex-GRCh38-2024-A --fastqs=/home/kim89/Cell_ranger/fastq_output/ --sample=SRR3879601,SRR3879602,SRR3879603 --localcores=8 --localmem=64 ERROR: No input FASTQs were found for the requested parameters. If your files came from bcl2fastq or mkfastq: - Make sure you are specifying the correct --sample(s), i.e. matching the sample sheet - Make sure your files follow the correct naming convention, e.g. SampleName_S1_L001_R1_001.fastq.gz (and the R2 version) - Make sure your --fastqs points to the correct location. - Make sure your --lanes, if any, are correctly specified. Refer to the "Specifying Input FASTQs" page at https://support.10xgenomics.com/ for more details.
- cellranger는 특정한 명명 규칙을 요구합니다. 파일 이름이 SampleName_S1_L001_R1_001.fastq.gz와 같은 형식이어야 샘플의 파일을 올바르게 식별할 수 있습니다.
- 파일 이름이 _R1_과 _R2_를 포함해야 PAIRED-END 데이터를 인식할 수 있습니다.
-
<SampleName> - An identifier for the sample, this is what Cell Ranger uses to determine which fastq files to combine into a single sample.
<SampleNumber> - This is the sample number based on the order that samples were listed in the sample sheet used when running bcl2fastq. This is not important for Cell Ranger, other than it indicates the end of the Sample Name, you can set all your samples to S1.
<Lane> - The lane number. If your sequencing was run across multiple lanes, then you may have multiple sets of fastqs for a single sample with different lane numbers.
<Read> - The read type: R1 for Read 1, R2 for Read 2, and index reads are I1 and I2.
001 - The last segment is always 001.
<SampleName>_S<SampleNumber>_L00<Lane>_<Read>_001.fastq.gz
-
mv 기존파일명 새로운파일명
mv SRR3879601_1.fastq.gz SRR3879601_S1_L001_R1_001.fastq.gz
mv SRR3879601_2.fastq.gz SRR3879601_S1_L001_R2_001.fastq.gz
mv SRR3879602_1.fastq.gz SRR3879602_S1_L001_R1_001.fastq.gz
mv SRR3879602_2.fastq.gz SRR3879602_S1_L001_R2_001.fastq.gz
mv SRR3879603_1.fastq.gz SRR3879603_S1_L001_R1_001.fastq.gz
mv SRR3879603_2.fastq.gz SRR3879603_S1_L001_R2_001.fastq.gz
+) 시컨싱 화학 명시 에러...(?) ⇨ 해결 방법 모르겠다...
2024-11-07 16:40:45 [runtime] (ready) ID.sample_1_analysis.SC_RNA_COUNTER_CS.SC_MULTI_CORE.MULTI_GEM_WELL_PROCESSOR.VDJ_T_GEM_WELL_PROCESSOR.SC_VDJ_CONTIG_ASSEMBLER.RUST_BRIDGE
2024-11-07 16:40:45 [runtime] (ready) ID.sample_1_analysis.SC_RNA_COUNTER_CS.SC_MULTI_CORE.MULTI_GEM_WELL_PROCESSOR.VDJ_T_GD_GEM_WELL_PROCESSOR.SC_VDJ_CONTIG_ASSEMBLER.RUST_BRIDGE
2024-11-07 16:40:45 [runtime] (failed) ID.sample_1_analysis.SC_RNA_COUNTER_CS.SC_MULTI_CORE.MULTI_CHEMISTRY_DETECTOR._GEM_WELL_CHEMISTRY_DETECTOR.DETECT_COUNT_CHEMISTRY
[error] Pipestance failed. Error log at:
sample_1_analysis/SC_RNA_COUNTER_CS/SC_MULTI_CORE/MULTI_CHEMISTRY_DETECTOR/_GEM_WELL_CHEMISTRY_DETECTOR/DETECT_COUNT_CHEMISTRY/fork0/chnk0-u2dee2c6efd/_errors
Log message:
We detected an unsupported chemistry combination (SC5P-R2, SC5P-PE). To process this combination of data you will need to specify SC5P-R2 via the --chemistry argument.
Waiting 6 seconds for UI to do final refresh.
Pipestance failed. Use --noexit option to keep UI running after failure.
2024-11-07 16:40:51 Shutting down.
Saving pipestance info to "sample_1_analysis/sample_1_analysis.mri.tgz"
For assistance, upload this file to 10x Genomics by running:
cellranger upload <your_email> "sample_1_analysis/sample_1_analysis.mri.tgz"이 오류 메시지는 cellranger가 샘플의 시퀀싱 화학(chemistry)을 감지했지만, 그 조합이 기본적으로 지원되지 않는다는 것을 의미합니다. SC5P-R2와 SC5P-PE 시퀀싱 화학 조합이 감지되었으며, 이를 처리하려면 --chemistry 인수를 사용하여 시퀀싱 화학을 명시적으로 지정해야 합니다.
cellranger count 명령어에 --chemistry=SC5P-R2 옵션을 추가하여 시퀀싱 화학을 명시적으로 지정하세요.- 해결안됨 !!! (전체 데이터를 다 안 가져오고 일부 데이터로만 실행해서 그런 거 같기도...)
cellranger count --id=sample1_analysis \
--transcriptome=/home/kim89/Cell_ranger/refdata-gex-GRCh38-2024-A \
--fastqs=/home/kim89/Cell_ranger/fastq_output/ \
--sample=SRR3879601,SRR3879602,SRR3879603 \
--chemistry=SC5P-R2 \
--localcores=8 --localmem=64< 참고 >
https://partrita.github.io/posts/scRNA-rawdata/
scRNA데이터 전처리하기
시작하기 앞서¶다음과 같은 몇가지 가정을 하고 시작하겠습니다. 가정1. 10x Genomics Single Cell Solution을 통해 얻은 서열 데이터를 사용 가정2. 리눅스(Ubuntu 22.04.3 LTS x86_64) OS를 사용하며 기본적인 she
partrita.github.io
Introduction to single-cell RNA-seq analysis
<!-- # Alignment and feature counting {#AliFeatCountTop} --> Introduction The first step in the analysis of single cell RNAseq data is to align the sequenced reads against a genomic reference and then use a transcriptome annotation to generate read counts
bioinformatics-core-shared-training.github.io
< 추가 >
https://blog.naver.com/shimyj123/222077459407?trackingCode=rss
'AI, 논문, 데이터 분석' 카테고리의 다른 글
| Batch Effect, Batch Correction (0) | 2024.11.21 |
|---|---|
| RNA 데이터 생성 과정 (0) | 2024.11.20 |
| SRA Toolkit 사용해서 데이터 받기 (3) | 2024.11.06 |
| Gene set enrichment analysis (GSEA) 논문 리뷰 (1) | 2024.10.11 |
| Gene Ontology (GO) Enrichment analysis 개념 정리 (1) | 2024.10.10 |

