먼저, Seurat과 관련된 library 설치
( Seurat 설치 에러는 아래 글 참고)
https://doraemin.tistory.com/36
[Seurat] 설치 ( + R 버전 에러)
Seurat 패키지 설치하기Seurat 공식 사이트의 'install' 부분의 코드 작성# Enter commands in R (or R studio, if installed)install.packages('Seurat')library(Seurat) https://satijalab.org/seurat/ Tools for Single Cell GenomicsA toolkit fo
doraemin.tistory.com
# Enter commands in R (or R studio, if installed)
#install.packages('Seurat')
library(Seurat) # scRNAseq
#install.packages('dplyr')
library(dplyr) # 데이터 집계/처리용
library(patchwork) # 그래프 그리기
#install.packages('ggplot2')
library(ggplot2)

if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("MAST")
BiocManager::install("limma")
BiocManager::install("DESeq2")







DEGs identification
We used a single-cell toolkit to pinpoint the DEG between D26 and D54 time points. The expression matrix was imported and converted into a Seurat object using the function ‘CreateSeuratObject’ from the Seurat package (Seurat v3.0). Then, quality control, normalization, feature selection, and clustering of marker genes of cells from scRNA-seq data were performed. We filtered out the cells with unique feature counts exceeding 10 000 or falling below 200 (Figure S1, see Supplementary Data are available online at http://bib.oxfordjournals.org/.).

DEG 식별
우리는 단일 세포 툴킷을 사용하여 D26과 D54 시점 사이의 DEG를 정확히 찾아냈습니다. 발현 행렬은 Seurat 패키지(Seurat v3.0)의 'CreateSeuratObject' 함수를 사용하여 Seurat 객체로 가져와 변환했습니다. 그런 다음 scRNA-seq 데이터에서 세포의 마커 유전자에 대한 품질 관리, 정규화, 특징 선택 및 클러스터링을 수행했습니다. 우리는 고유한 특징 수가 10,000을 초과하거나 200 미만인 세포를 필터링했습니다(그림 S1 , 보충 자료는 http://bib.oxfordjournals.org/ 에서 온라인으로 볼 수 있습니다.).
# Replace with the actual path to your TPM data
tpm_data <- read.csv("C:\\Users\\LG\\Documents\\MJU\\연구실\\24.09.20_scRNA 데이터 분석\\GSE86982_smartseq.tpm.csv", row.names = 1, header = TRUE)
# Check the row names or metadata to identify stages
head(rownames(tpm_data)) # View the row names to check for stage information
# Check the column names to view the stage information
head(colnames(tpm_data)) # This should show identifiers related to D26, D54, and other stages
# "26" 또는 "54"가 포함된 열 이름을 찾는 경우
d26_d54_cells <- grepl("26|54", colnames(tpm_data))
tpm_d26_d54 <- tpm_data[, d26_d54_cells] # 해당 세포만 포함한 데이터로 서브셋 만들기
# Seurat 객체 생성 (D26과 D54 세포만 포함)
seurat_object <- CreateSeuratObject(counts = as.matrix(tpm_d26_d54), project = "TPM_D26_D54", min.cells = 3, min.features = 200)

이 경고 메시지들은 심각한 오류가 아니며, 데이터를 처리하는 과정에서 자동으로 처리되는 부분입니다.
1. 경고: Feature names cannot have underscores ('_'), replacing with dashes ('-')
- 이 경고는 Seurat에서 피처 이름(유전자 이름 등)에 밑줄(_)을 사용할 수 없기 때문에 이를 대시(-)로 자동 변경했다는 내용입니다.
- 그러나 피처 이름에 대해 수동으로 변경하고 싶다면 다음과 같은 코드를 사용하여 미리 처리할 수 있습니다:
rownames(tpm_matrix) <- gsub("_", "-", rownames(tpm_matrix))2. 경고: Data is of class matrix. Coercing to dgCMatrix.
- 이 경고는 입력 데이터 tpm_matrix가 matrix 클래스인 반면, Seurat에서는 희소 행렬 형식인 dgCMatrix 클래스를 선호하기 때문에 자동으로 변환되었다는 의미입니다.
############### Seurat객체의 QC 전후, 바이올린 플랏 ### 퀄리티가 안 좋은 세포들은 버리자.
# 메타데이터 추가: nFeature_RNA, nCount_RNA, percent.mt 계산
# 미토콘드리아 유전자 비율 계산 (MT로 시작하는 유전자 이름 패턴 사용)
seurat_object[["percent.mt"]] <- PercentageFeatureSet(seurat_object, pattern = "^MT")
# 세포 이름에서 D26 또는 D54 라벨 추가 (메타데이터에 추가)
seurat_object$stage <- ifelse(grepl("26D", colnames(seurat_object)), "D26", "D54")
# 그룹별 바이올린 플롯 그리기
VlnPlot(seurat_object, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), group.by = "stage", ncol = 3)
# D26 세포만 필터링
d26_subset <- subset(seurat_object, subset = stage == "D26")
d26_subset$stage <- "D26"
# D26 세포에 대해 그룹화된 바이올린 플롯 그리기
VlnPlot(d26_subset, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), group.by = "stage", ncol = 3) +
ylim(0, 10000) + # nFeature_RNA 범위 설정
ylim(0, 2000000) + # nCount_RNA 범위 설정
ylim(0,20) # percent.mt 범위 설정

# QC 기준 적용
seurat_object_qc <- subset(seurat_object, subset = nFeature_RNA > 200 & nFeature_RNA < 10000 & percent.mt < 5)
# QC 적용 후 바이올린 플롯 (y축 범위 설정)
VlnPlot(seurat_object_qc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), group.by = "stage", ncol = 3)
#+ ylim(0, 10000) + # nFeature_RNA 범위
# ylim(0, 2000000) + # nCount_RNA 범위
# ylim(0, 80) # percent.mt 범위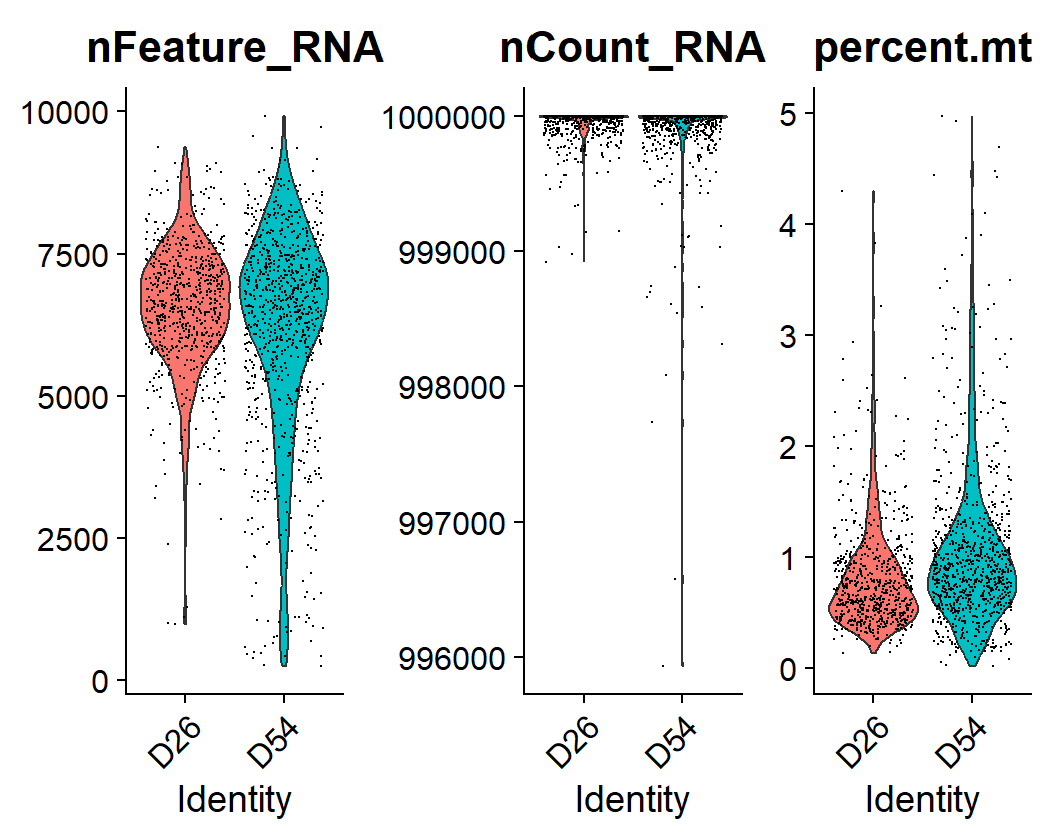
We also removed cells with mitochondrial and ribosomal counts greater than 20%. The ‘LogNormalize’ global-scaling normalization method was used to standardise individual cells’ feature expression measurements. Then, selection.method = ‘vst’ was used to find feature variables. Each gene's expression level was normalized by considering the total number of unique molecular identifiers (UMI) within each cell, followed by applying a natural logarithm to the UMI counts.
또한 미토콘드리아와 리보솜 수가 20% 이상인 세포도 제거했습니다. 'LogNormalize' 전역 스케일링 정규화 방법을 사용하여 개별 세포의 특성 발현 측정을 표준화했습니다. 그런 다음 selection.method = 'vst'를 사용하여 특성 변수를 찾았습니다. 각 유전자의 발현 수준은 각 세포 내의 총 고유 분자 식별자(UMI) 수를 고려하여 정규화한 다음 UMI 수에 자연 대수를 적용했습니다.
# Preprocessing :
# 미토콘드리아 유전자 비율(percent.mt) 계산 (이미 처리된 부분)
seurat_object[["percent.mt"]] <- PercentageFeatureSet(seurat_object, pattern = "^MT-")
# 리보솜 유전자 비율(percent.rb) 계산
seurat_object[["percent.rb"]] <- PercentageFeatureSet(seurat_object, pattern = "^RPS|^RPL")
# 미토콘드리아와 리보솜 수가 20% 이상인 세포 필터링
seurat_object <- subset(seurat_object, subset = percent.mt < 20 & percent.rb < 20)
# 데이터 정규화 (LogNormalize 방식 사용)
seurat_object <- NormalizeData(seurat_object, normalization.method = "LogNormalize", scale.factor = 10000)
# 가변적 특성(변동성이 큰 유전자) 찾기
seurat_object <- FindVariableFeatures(seurat_object, selection.method = "vst", nfeatures = 2000)
# 데이터 스케일링
seurat_object <- ScaleData(seurat_object)설명:
- 미토콘드리아 필터링: seurat_object[["percent.mt"]]는 미토콘드리아 유전자의 발현 비율을 계산하는 코드입니다. MT로 시작하는 유전자를 기준으로 비율을 계산합니다.
- 리보솜 필터링: seurat_object[["percent.rb"]]를 사용해 리보솜 유전자(RPS와 RPL로 시작하는 유전자)의 비율을 계산했습니다.
- 세포 필터링: 미토콘드리아 유전자 또는 리보솜 유전자의 발현 비율이 20%를 초과하는 세포는 제거했습니다.
- LogNormalize 방식 정규화: LogNormalize는 각 세포의 총 UMI 수에 따라 각 유전자의 발현 수준을 정규화한 후 자연 대수를 적용하는 방식입니다. 이 방법은 이미 UMI 수를 기반으로 한 자연 대수 적용을 포함하고 있으므로 별도로 추가할 필요는 없습니다.




# D26과 D54 세포에 대한 정확한 조건 설정
seurat_object$stage <- ifelse(grepl("26D", colnames(tpm_d26_d54)), "D26",
ifelse(grepl("54", colnames(tpm_d26_d54)), "D54", NA))
# stage 메타데이터의 값을 확인
head(seurat_object$stage)
# stage 메타데이터를 ident으로 설정
Idents(seurat_object) <- seurat_object$stage
# 현재 정체성 확인
levels(Idents(seurat_object))
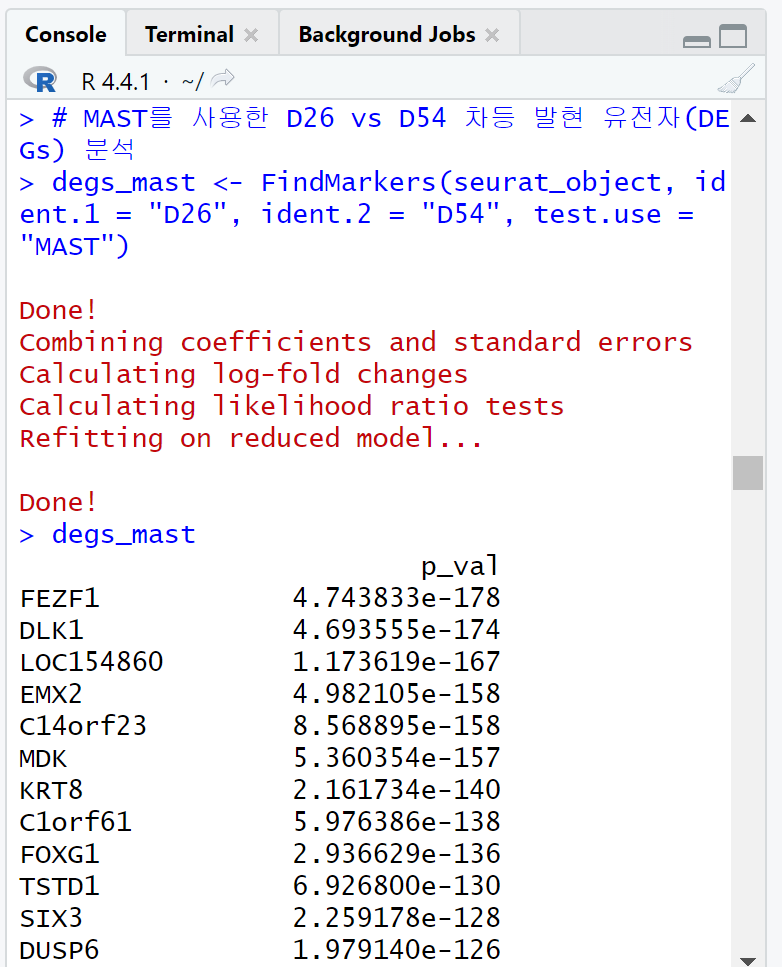
# MAST를 사용한 D26 vs D54 차등 발현 유전자(DEGs) 분석
degs_mast <- FindMarkers(seurat_object, ident.1 = "D26", ident.2 = "D54", test.use = "MAST")

'AI, 논문, 데이터 분석' 카테고리의 다른 글
| [Seurat] [limma] DEG 분석 (0) | 2024.09.19 |
|---|---|
| [Seurat] [MAST] DEGs 분석 (0) | 2024.09.19 |
| [Seurat] 설치 ( + R 버전 에러) (0) | 2024.09.18 |
| read alignment STAR (0) | 2024.09.05 |
| SmartSeq2(Smart Sequencing Technology 2) (1) | 2024.09.05 |



