1. Linux 환경에서 설치 밀 실행해 보았다.
터미널에서, conda를 사용하여, Scanpy 패키지 설치
conda install -c conda-forge scanpy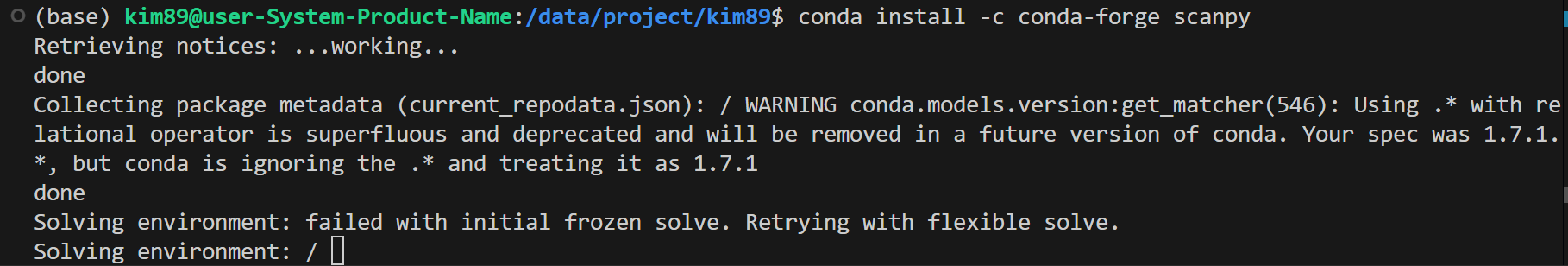
더보기
가상환경 실행 후, (가상환경에서) 설치




(약 1시간 정도? 다운로드가 이어지다가, Proceed 물으면) Proceed 응답 눌러주기
EnvironmentNotWritableError가 나타났습니다. 이는 현재 사용자가 /packages/anaconda3 경로에 대한 쓰기 권한이 없음을 의미합니다.
그래서, conda 가상환경에서 실행해보자.
미리 만들어준 가상환경 활성화하고, Scanpy 패키지 설치.
conda activate kim89_env
conda install -c conda-forge scanpy(가상환경에서) Scanpy 설치 완료!
2. Scanpy 설치 확인
설치 후 터미널에서 Python 인터프리터를 실행하고 Scanpy를 불러오기
python
>>> import scanpy as sc
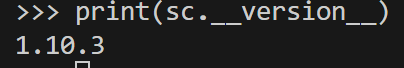
>>> print(sc.__version__) # Scanpy 버전 출력
이제 python에서 Scanpy를 활용한 데이터 분석을 진행해보자.
1. 데이터 로딩
먼저, 터미널에서 python 실행
python
이제, python 코드를 작성.
import scanpy as sc
adata = sc.read_10x_mtx(
path="/실제경로/작성", # matrix.mtx 파일이 있는 경로
var_names='gene_symbols', # features.tsv에서 두 번째 열(유전자 심볼) 사용
cache=True # 데이터를 캐싱하여 빠르게 재사용
)
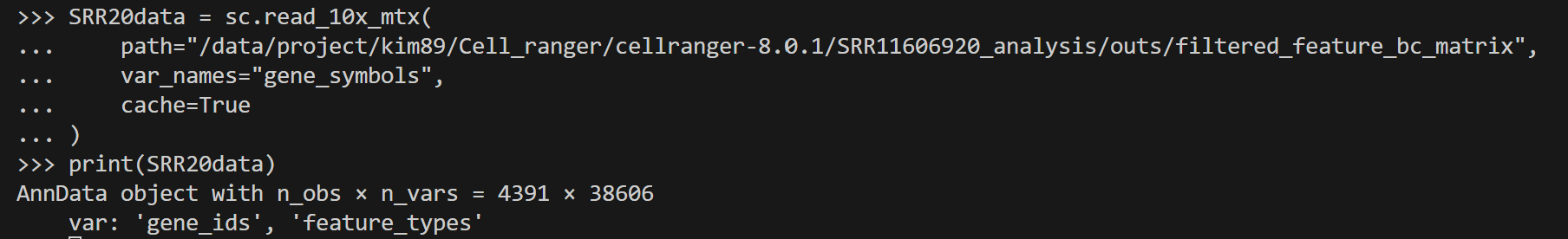
SRR20data = sc.read_10x_mtx(
path="/data/project/kim89/Cell_ranger/cellranger-8.0.1/SRR11606920_analysis/outs/filtered_feature_bc_matrix",
var_names="gene_symbols",
cache=True
)
print(SRR20data)
2. QC 및 필터링, 정규화 및 변동 유전자 선택
python 파일을 따로 만들어주었다.
import scanpy as sc
import harmonypy as hm
import pandas as pd
# 1. 데이터 로드
# 10개의 데이터 파일 경로 지정
file_paths = [
"/data/project/kim89/Cell_ranger/cellranger-8.0.1/SRR11606920_analysis/outs/filtered_feature_bc_matrix",
"/data/project/kim89/Cell_ranger/cellranger-8.0.1/SRR11606921_analysis/outs/filtered_feature_bc_matrix",
"/data/project/kim89/Cell_ranger/cellranger-8.0.1/SRR11606922_analysis/outs/filtered_feature_bc_matrix",
"/data/project/kim89/Cell_ranger/cellranger-8.0.1/SRR11606923_analysis/outs/filtered_feature_bc_matrix",
"/data/project/kim89/Cell_ranger/cellranger-8.0.1/SRR11606924_analysis/outs/filtered_feature_bc_matrix",
"/data/project/kim89/Cell_ranger/cellranger-8.0.1/SRR11606925_analysis/outs/filtered_feature_bc_matrix",
"/data/project/kim89/Cell_ranger/cellranger-8.0.1/SRR11606926_analysis/outs/filtered_feature_bc_matrix",
"/data/project/kim89/Cell_ranger/cellranger-8.0.1/SRR11606927_analysis/outs/filtered_feature_bc_matrix",
"/data/project/kim89/Cell_ranger/cellranger-8.0.1/SRR11606928_analysis/outs/filtered_feature_bc_matrix",
"/data/project/kim89/Cell_ranger/cellranger-8.0.1/SRR11606929_analysis/outs/filtered_feature_bc_matrix"
]
# 각 데이터를 Scanpy의 AnnData로 읽어들임
adatas = [sc.read_10x_mtx(path, var_names="gene_symbols", cache=True) for path in file_paths]
# donor 정보 추가
# donor ID 리스트
donor_ids = [
"donor_1", "donor_2", "donor_3",
"donor_4_1", "donor_5_1",
"donor_4_2", "donor_5_2",
"donor_6", "donor_7", "donor_8"
]
# 질병 상태 리스트 (normal/thrombocytopenia)
disease_states = [
"normal", "normal", "normal",
"thrombocytopenia", "thrombocytopenia",
"normal", "normal",
"thrombocytopenia", "thrombocytopenia", "thrombocytopenia"
]
# 각 데이터에 donor ID를 추가
# for adata, donor_id in zip(adatas, donor_ids):
# adata.obs["batch"] = donor_id
# 각 데이터에 donor ID와 disease 상태를 추가
for adata, donor_id, disease in zip(adatas, donor_ids, disease_states):
adata.obs["batch"] = donor_id
adata.obs["disease"] = disease
# 데이터 확인
# for adata in adatas:
# print(adata.obs["batch"].unique())
# 2. 데이터 결합
# 공통 유전자 기준으로 데이터를 결합
# 모든 데이터를 결합
merged_adata = adatas[0].concatenate(
*adatas[1:], batch_key="batch", batch_categories=donor_ids
)
# disease 정보 추가
merged_adata.obs["disease"] = pd.concat([adata.obs["disease"] for adata in adatas]).values
# 결합된 데이터의 batch 확인
# print(merged_adata.obs["batch"].unique())
print("Merged data:", merged_adata)
"""
Merged data: AnnData object with n_obs × n_vars = 37686 × 38606
obs: 'batch', 'disease'
var: 'gene_ids', 'feature_types'
"""
# 3. QC (필터링 전 기본 QC 메트릭 계산)
# 미토콘드리아 유전자 필터링
merged_adata.var["mt"] = merged_adata.var_names.str.startswith("MT-")
sc.pp.calculate_qc_metrics(merged_adata, qc_vars=["mt"], inplace=True)
# before QC plot
sc.settings.figdir = "/data/project/kim89/SRR116069__plots"
sc.pl.violin(
merged_adata,
keys=["total_counts", "n_genes_by_counts", "pct_counts_mt"],
jitter=0.4,
multi_panel=True,
save="plot_QC_before.png"
)
# QC 필터링 기준 설정 (Fan 기준 사용)
QC_filter = (
(merged_adata.obs["n_genes_by_counts"] > 200) & # 유전자 수 > 200
(merged_adata.obs["n_genes_by_counts"] < 2500) & # 유전자 수 < 2500
(merged_adata.obs["total_counts"] > 300) & # UMI > 300
(merged_adata.obs["total_counts"] < 15000) & # UMI < 15000
(merged_adata.obs["pct_counts_mt"] < 10) # 미토콘드리아 비율 < 10%
)
# 필터링 적용
filtered_adata = merged_adata[QC_filter, :]
print("Filtered data:", filtered_adata)
"""
Filtered data: View of AnnData object with n_obs × n_vars = 33062 × 38606
obs: 'batch', 'disease', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'feature_types', 'mt', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts'
"""
# after QC plot
# # Fan 기준
sc.pl.violin(
filtered_adata,
keys=["total_counts", "n_genes_by_counts", "pct_counts_mt"],
jitter=0.4,
multi_panel=True,
save="plot_QC_after.png"
)
# 필터링된 데이터 저장 (.h5ad 형식으로 저장하여 이후 분석에 사용)
# filtered_adata.write_h5ad("filtered_adata.h5ad")
# 정규화 전 데이터의 총 카운트 분포 확인
# 33062개의 세포, 38606개의 유전자
# total_counts의 평균: 약 3047 # 분포가 세포마다 다르고, 최대값이 약 14487로 세포 간 차이가 큼.
print("정규화 전:")
print(filtered_adata.obs["total_counts"].describe())
"""
정규화 전:
count 33062.000000
mean 3047.830322
std 1660.008179
min 500.000000
25% 1942.000000
50% 2775.000000
75% 3787.000000
max 14487.000000
Name: total_counts, dtype: float64
"""
# 정규화 전 첫 번째 세포의 값
print("정규화 전 첫 번째 세포:")
print(merged_adata.X[0, :])
"""
정규화 전 첫 번째 세포:
<Compressed Sparse Row sparse matrix of dtype 'float32'
with 1290 stored elements and shape (1, 38606)>
Coords Values
(0, 58) 1.0
(0, 81) 1.0
...
"""
# 4. 정규화
# 총 발현값으로 정규화
sc.pp.normalize_total(filtered_adata, target_sum=1e4)
# 로그 변환
sc.pp.log1p(filtered_adata)
# 변수 유전자 선택
sc.pp.highly_variable_genes(filtered_adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
filtered_adata = filtered_adata[:, filtered_adata.var.highly_variable]
# 데이터 확인
# 정규화 후 총합 다시 계산
sc.pp.calculate_qc_metrics(filtered_adata, inplace=True) ###########################################해야할지 말아야할지,,
# 정규화 후 데이터의 총 카운트 분포 확인
# 33062개의 세포, 3052개의 변수 유전자 (38606개의 유전자에서 줄어듦)
# total_counts의 평균: 약 272 # 모든 세포가 동일한 총합(target_sum)에 맞추어 정규화되었으며, 값의 범위가 좁아짐.
print("정규화 후:")
print(filtered_adata.obs["total_counts"].describe())
"""
정규화 후:
count 33062.000000
mean 272.048615
std 89.915779
min 40.955204
25% 214.527405
50% 265.780319
75% 319.822426
max 1039.758545
Name: total_counts, dtype: float64
"""
# 정규화 후 첫 번째 세포의 값
print("정규화 후 첫 번째 세포:")
print(filtered_adata.X[0, :])
"""
정규화 후 첫 번째 세포:
<Compressed Sparse Row sparse matrix of dtype 'float32'
with 196 stored elements and shape (1, 3052)>
Coords Values
(0, 8) 1.6668635606765747
(0, 26) 2.2608320713043213
...
"""
print(filtered_adata)
"""
View of AnnData object with n_obs × n_vars = 33062 × 3052
obs: 'batch', 'disease', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'feature_types', 'mt', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'log1p', 'hvg'
"""
# normalization plot
sc.pl.violin(
filtered_adata,
keys=["total_counts", "n_genes_by_counts", "pct_counts_mt"],
jitter=0.4,
multi_panel=True,
save="plot_normalization.png"
)
# 5. 배치 효과 조정 (Harmony 사용), PCA, 클러스터링
import harmonypy as hm
# PCA 계산
sc.tl.pca(filtered_adata)
print(filtered_adata.obsm['X_pca'].shape) # (셀 수, PCA 차원 수) : (33062, 50)
# Harmony 실행
# harmony_results = hm.run_harmony(
# filtered_adata.obsm['X_pca'], # PCA 결과
# filtered_adata.obs, # 관측값 데이터 (obs) (셀 수, 메타데이터 열 수)
# 'batch' # 배치 효과를 조정할 열 이름
# )
sc.tl.pca(filtered_adata)
harmony_results = hm.run_harmony(
filtered_adata.obsm['X_pca'],
filtered_adata.obs,
['batch', 'disease'] # 배치와 질병 상태를 함께 조정
)
# Harmony 결과 확인
print("Before transpose:", harmony_results.Z_corr.shape) # (50, 33062)
# Z_corr는 (PCA 차원, 셀 수) 형식으로 반환되므로 이를 Transpose(전치)하여 (셀 수, PCA 차원) 형식으로 맞춰야 합니다.
# Transpose하여 저장
filtered_adata.obsm['X_pca_harmony'] = harmony_results.Z_corr.T
print("After transpose, Harmony 결과 저장 완료:", filtered_adata.obsm['X_pca_harmony'].shape) # (33062, 50)
# Neighbors 계산
sc.pp.neighbors(filtered_adata, use_rep="X_pca_harmony")
print("Neighbors 계산 완료")
# UMAP 계산
sc.tl.umap(filtered_adata)
print("UMAP 계산 완료")
# Leiden 클러스터링 수행
sc.tl.leiden(filtered_adata, resolution=1.0) # resolution 값 조정 가능
print("Leiden 클러스터링 결과:", filtered_adata.obs['leiden'].value_counts())
"""
Leiden 클러스터링 결과: leiden
0 5493
1 4494
2 4470
3 4251
4 2754
5 2737
6 2088
7 1813
8 1505
9 1113
10 977
11 949
12 175
13 165
14 42
15 36
Name: count, dtype: int64
"""
# UMAP 시각화
# sc.pl.umap(filtered_adata, color=["batch", "leiden"], save="batch_effect_umap.png")
sc.pl.umap(filtered_adata, color=["batch", "disease", "leiden"], save="donor_and_disease.png")
print("UMAP 시각화 완료")
# 데이터 저장
# filtered_adata.write_h5ad("merged_filtered_normalized_batch_corrected.h5ad")
print("데이터 저장 완료")
############# << 3. 탐색분석 >> ###########
# 0. DEGs
# 클러스터 간 차이를 확인하기 위해 주요 차별 발현 유전자(DEGs)를 식별
sc.tl.rank_genes_groups(filtered_adata, 'leiden', method='t-test')
# sc.pl.rank_genes_groups(filtered_adata, n_genes=20, save="ranked_genes.png")
sc.pl.rank_genes_groups(filtered_adata, n_genes=20, save="ranked_genes_with_disease.png")
# 클러스터별 UMAP 시각화
sc.pl.umap(filtered_adata, color=["leiden", "LYZ"], save="gene_highlight_LYZ.png") ##### 특정 유전자 NKG7의 cluster 존재여부
# 1. 기능 강화 분석 (Functional Enrichment Analysis)
# 생물학적 기능적 특성을 도출할 수 있습니다. 이를 위해 Gene Ontology (GO), KEGG Pathway 분석, 그리고 GSVA를 사용할 수 있습니다.
# (1) 주요 유전자 집합 준비
# 주요 유전자 추출
import pandas as pd
top_genes = pd.DataFrame(filtered_adata.uns['rank_genes_groups']['names']).head(50) # 상위 50개 유전자
# print(top_genes)
# 0 1 2 ... 14 15 16
# 0 LEF1 LYZ NKG7 ... GNLY CD74 HSP90AA1
# 1 CAMK4 CST3 GNLY ... NKG7 AFF3 NKG7
# [50 rows x 17 columns]
# 클러스터 0의 상위 유전자 추출
cluster_0_genes = filtered_adata.uns['rank_genes_groups']['names']['0'][:50] # 클러스터 0의 상위 50개 유전자
# print(cluster_0_genes)
# ['LEF1' 'CAMK4' 'TSHZ2' 'PRKCA' 'INPP4B' 'PDE3B' 'SERINC5' 'BCL11B' 'FHIT'
# 'NELL2' 'BACH2' 'TXK' 'BCL2' 'ANK3' 'ENSG00000271774' 'TC2N' 'TRABD2A'
# 'PLCL1' 'ENSG00000290067' 'MLLT3' 'ENSG00000249806' 'LNCRNA-IUR' 'SESN3'
# 'LINC01550' 'IGF1R' 'THEMIS' 'PATJ' 'PRKCA-AS1' 'ENSG00000253980' 'NR3C2'
# 'LINC02964' 'RETREG1' 'CSGALNACT1' 'APBA2' 'EDA' 'ABLIM1' 'ZC3H12B' 'AK5'
# 'ITGA6' 'RASGRF2' 'IMMP2L' 'ANKRD55' 'PRKN' 'LEF1-AS1' 'ICOS' 'KANK1'
# 'MDS2' 'ZEB1' 'ENSG00000286330' 'FAAH2']
# print(filtered_adata.uns['rank_genes_groups']['names']['0'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['1'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['2'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['3'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['4'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['5'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['6'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['7'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['8'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['9'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['10'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['11'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['12'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['13'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['14'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['15'][:10])
# print(filtered_adata.uns['rank_genes_groups']['names']['16'][:10])
# (2) GO 및 KEGG Pathway 분석
# gprofiler-official를 사용한 분석
from gprofiler import GProfiler
# GO 및 KEGG 분석
gp = GProfiler(return_dataframe=True)
# 클러스터 0의 상위 유전자 리스트로 변환
cluster_0_genes_list = list(set(cluster_0_genes.tolist())) # 중복 제거 포함
print(type(cluster_0_genes)) # <class 'numpy.ndarray'>
print(type(cluster_0_genes.tolist())) # <class 'list'>
# gProfiler 분석
go_results = gp.profile(organism='hsapiens', query=cluster_0_genes_list) ##### cluster 0에서의 GO
# print(go_results.head()) # 상위 몇 줄 출력
# source native name ... recall query parents
# 0 GO:BP GO:0048856 anatomical structure development ... 0.004238 query_1 [GO:0032502]
# 1 GO:BP GO:0032502 developmental process ... 0.004029 query_1 [GO:0008150]
# 2 GO:BP GO:0010646 regulation of cell communication ... 0.005518 query_1 [GO:0007154, GO:0050794]
# 3 GO:BP GO:0007275 multicellular organism development ... 0.004523 query_1 [GO:0032501, GO:0048856]
# 4 GO:BP GO:0009653 anatomical structure morphogenesis ... 0.005961 query_1 [GO:0032502, GO:0048856]
# [5 rows x 14 columns]
# print(go_results.columns) # 열 이름 확인
# Index(['source', 'native', 'name', 'p_value', 'significant', 'description',
# 'term_size', 'query_size', 'intersection_size', 'effective_domain_size',
# 'precision', 'recall', 'query', 'parents'],
# dtype='object')
print(go_results[['name', 'p_value', 'source']]) # 결과 확인
# 저장
go_results.to_csv("GO_KEGG_enrichment_results.csv", index=False)
# import matplotlib.pyplot as plt
# import seaborn as sns
# import numpy as np
# # p-value를 기준으로 상위 10개 경로 시각화
# top_results = go_results.nsmallest(10, 'p_value')
# sns.barplot(y=top_results['native'], x=-np.log10(top_results['p_value']))
# plt.xlabel('-log10(p-value)')
# plt.ylabel('GO Terms / KEGG Pathways')
# plt.show()
# from pyGSVA import gsva
# # Gene set 정의 (MSigDB 또는 KEGG)
# gene_sets = {
# "Apoptosis": ["TP53", "BCL2", "CASP3"], # 예시 유전자
# "Cell_Cycle": ["CDK1", "CDK2", "CCNB1"]
# }
# # GSVA 분석
# gsva_results = gsva(expression_data=filtered_adata.to_df(), gene_sets=gene_sets, method='gsva')
# print(gsva_results)
##########
# 2. 전사인자 분석 (Transcription Factor Analysis)
import scanpy as sc
from pyscenic.pipeline import run_pyscenic_pipeline
# 데이터 준비
filtered_adata.raw = filtered_adata # raw data 저장 (pySCENIC은 raw counts 필요)
filtered_adata.X = filtered_adata.raw.to_adata().X # raw counts 사용
# pySCENIC 실행
run_pyscenic_pipeline(
adata=filtered_adata,
output_prefix='pyscenic_results',
organism='homo_sapiens',
dbs=['cisTarget_human_v10_clust.genes_vs_motifs.rankings.db'],
n_threads=4
)
# 결과 파일 읽기
import pandas as pd
regulons = pd.read_csv('pyscenic_results/regulons.csv')
# 상위 전사인자 시각화
sc.pl.dotplot(filtered_adata, var_names=regulons.head(20), groupby='leiden')
# # 3. 의사시간 분석 (Pseudotime Analysis)
# # 의사시간 계산
# sc.tl.dpt(filtered_adata, n_dcs=10) # DPT 계산
# sc.pl.umap(filtered_adata, color='dpt_pseudotime', save="pseudotime.png")
# # 4. 세포 간 상호작용 (Cell-Cell Interaction Analysis)
# import squidpy as sq
# # Spatial data 준비 (spatial data가 없을 경우 random assignment 가능)
# adata = sq.datasets.imc() # 예제 데이터
# # Ligand-Receptor 상호작용 계산
# sq.gr.ligrec(adata)
# # 시각화
# sq.pl.ligrec(adata, target_groups="leiden", source_groups="batch", dendrogram=True)
# # 5.세포 주기 분석 (Cell Cycle Analysis)
# # 세포 주기 점수 계산
# s_genes = ["MCM5", "PCNA", "TYMS", "FEN1", "MCM2", "MCM4", "RRM1", "UNG"]
# g2m_genes = ["CCNB1", "CCNB2", "CDC20", "CDK1", "PLK1", "TOP2A"]
# sc.tl.score_genes_cell_cycle(filtered_adata, s_genes=s_genes, g2m_genes=g2m_genes)
# # UMAP 시각화
# sc.pl.umap(filtered_adata, color=["phase", "S_score", "G2M_score"], save="cell_cycle.png")
더보기
import scanpy as sc
# 1. 데이터 로드
SRR20data = sc.read_10x_mtx(
path="/data/project/kim89/Cell_ranger/cellranger-8.0.1/SRR11606920_analysis/outs/filtered_feature_bc_matrix",
var_names="gene_symbols",
cache=True
)
print(SRR20data)
"""
AnnData object with n_obs × n_vars = 4391 × 38606
var: 'gene_ids', 'feature_types'
"""
# 미토콘드리아 유전자 필터링 (유전자 이름에 "MT-"가 포함된 경우)
SRR20data.var["mt"] = SRR20data.var_names.str.startswith("MT-")
# 2. QC 메트릭 계산
sc.pp.calculate_qc_metrics(SRR20data, qc_vars=["mt"], percent_top=None, log1p=False, inplace=True)
# 3. QC 메트릭 확인
print(SRR20data.obs[["total_counts", "n_genes_by_counts", "pct_counts_mt"]].head())
"""
total_counts n_genes_by_counts pct_counts_mt
AAACCTGAGAACTCGG-1 2328.0 1290 3.221649
AAACCTGAGTTGCAGG-1 3041.0 1302 2.301874
AAACCTGCAAATCCGT-1 2202.0 999 3.451408
AAACCTGCAATGGATA-1 2108.0 988 3.462998
AAACCTGCAGGCTCAC-1 4166.0 1292 1.968315
"""
# 총 UMI 수와 유전자 수
sc.settings.figdir = "/data/project/kim89/plots"
# sc.pl.violin(
# SRR20data,
# keys=["total_counts", "n_genes_by_counts", "pct_counts_mt"],
# jitter=0.4,
# multi_panel=True,
# save="violin_plot_QC_before.png"
# )
# 4. 필터링 기준 설정
# Lambrechts 기준: ≤ 100 또는 ≥ 6000 유전자 / ≤ 200 UMI / ≥ 10% 미토콘드리아 비율
Lambrechts_filter = (
(SRR20data.obs["n_genes_by_counts"] > 100) &
(SRR20data.obs["n_genes_by_counts"] < 6000) &
(SRR20data.obs["total_counts"] > 200) &
(SRR20data.obs["pct_counts_mt"] < 10)
)
# Fan 기준: 200 < 유전자 수 < 2500 / 300 < UMI < 15000 / 미토콘드리아 비율 < 10%
Fan_filter = (
(SRR20data.obs["n_genes_by_counts"] > 200) &
(SRR20data.obs["n_genes_by_counts"] < 2500) &
(SRR20data.obs["total_counts"] > 300) &
(SRR20data.obs["total_counts"] < 15000) &
(SRR20data.obs["pct_counts_mt"] < 10)
)
# 5. 필터 적용 (원하는 기준을 선택)
# Lambrechts 기준
SRR20data_filtered_Lambrechts = SRR20data[Lambrechts_filter, :]
# Fan 기준
SRR20data_filtered_Fan = SRR20data[Fan_filter, :]
# 필터링 후 데이터 크기 확인
print("Lambrechts 기준 적용 후:", SRR20data_filtered_Lambrechts)
print("Fan 기준 적용 후:", SRR20data_filtered_Fan)
# # 6. 필터링 전/후 QC 시각화
# # Lambrechts 기준
# sc.pl.violin(
# SRR20data_filtered_Lambrechts,
# keys=["total_counts", "n_genes_by_counts", "pct_counts_mt"],
# jitter=0.4,
# multi_panel=True,
# save="violin_plot_Lambrechts_QC_after.png"
# )
# # Fan 기준
# sc.pl.violin(
# SRR20data_filtered_Fan,
# keys=["total_counts", "n_genes_by_counts", "pct_counts_mt"],
# jitter=0.4,
# multi_panel=True,
# save="violin_plot_Fan_QC_after.png"
# )
# 필터링된 데이터 저장 (.h5ad 형식으로 저장하여 이후 분석에 사용)
# SRR20data_filtered_Lambrechts.write_h5ad("SRR20data_filtered_Lambrechts.h5ad")
# SRR20data_filtered_Fan.write_h5ad("SRR20data_filtered_Fan.h5ad")
# 7. 정규화
# (1) 데이터를 복사하여 정규화 작업 시작
SRR20data_norm = SRR20data_filtered_Fan.copy()
# (2) 세포마다 총 발현값을 기준으로 정규화 (1e4 = 10,000으로 정규화)
sc.pp.normalize_total(SRR20data_norm, target_sum=1e4)
# (3) 로그 변환 적용
sc.pp.log1p(SRR20data_norm)
# (4) 변수 유전자 선택 (Highly Variable Genes)
sc.pp.highly_variable_genes(SRR20data_norm, min_mean=0.0125, max_mean=3, min_disp=0.5)
# 변수 유전자만 선택
SRR20data_norm = SRR20data_norm[:, SRR20data_norm.var.highly_variable]
# (5) 데이터 확인
# 정규화 후 총합 다시 계산
sc.pp.calculate_qc_metrics(SRR20data_norm, inplace=True)
# 정규화 전 데이터의 총 카운트 분포 확인
print("정규화 전:")
print(SRR20data_filtered_Fan.obs["total_counts"].describe())
# 정규화 후 데이터의 총 카운트 분포 확인
print("정규화 후:")
print(SRR20data_norm.obs["total_counts"].describe())
# 정규화 전 첫 번째 세포의 값
print("정규화 전 첫 번째 세포:")
print(SRR20data_filtered_Fan.X[0, :])
# 정규화 후 첫 번째 세포의 값
print("정규화 후 첫 번째 세포:")
print(SRR20data_norm.X[0, :])
print(SRR20data_norm)
"""
View of AnnData object with n_obs × n_vars = 4034 × 3781
obs: 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'feature_types', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'log1p', 'hvg'
"""
# plot
sc.pl.violin(
SRR20data_norm,
keys=["total_counts", "n_genes_by_counts", "pct_counts_mt"],
jitter=0.4,
multi_panel=True,
save="violin_plot_normalization.png"
)
# 정규화된 데이터를 저장
# SRR20data_norm.write_h5ad("SRR20data_normalized.h5ad")해당 파일을 실행
(kim89_env) kim89@user-System-Product-Name:/data/project/kim89$ python SRR116069__Scanpy.py'dev-setup' 카테고리의 다른 글
| [vllm] 설치하기 (Linux 환경을 위해, WSL 2 환경에서 설치) (0) | 2025.02.24 |
|---|---|
| [PyTorch] Anaconda, PyTorch 설치 및 실행 (1) | 2025.01.17 |
| conda 가상환경 생성 및 R 실행 (3) | 2024.11.28 |
| [Linux] Hard Link vs. Soft Link (2) | 2024.11.26 |
| Cell Ranger 설치 및 실행 (2) | 2024.11.07 |



